Outline of selected topics for
current and future research
Back to Home Page Related Jobs
Publications
To those interested in
collaborating with us: The
following description of current research projects and anticipated future
interests reveals a specific vision for the continuation and expansion of my
research program in atomic, molecular, optical, and surface (AMOS) physics at
KSU. Depending on the availability of funds, experimental advances, and
intriguing new ideas my group is always open towards new and/or collaborative
projects, in addition to the ones listed below.
To interested students: If
you would like to know more about our research or are interested in working in
my group, don’t hesitate to call or email me, or simply stop by my office. Whether you are new to research in
theoretical AMOS physics or already have experience, I will find a topic of
current research interest for you to work on.
Summary
In close collaboration with experimental groups and motivated in part
by available and emerging intense short-pulse laser and X-ray light sources, my
research group seeks to develop conceptual, analytical, and numerical tools for
the theoretical description of the
a) coherent electronic dynamics
in atoms and
b) coherent electronic and nuclear dynamics in molecules, clusters, and
solid surfaces.
This research includes a number of projects that are sketched below.
Their unifying theme is the study of dynamical processes at the natural time
scale of either the electronic motion (attoseconds, 1as=10-18s) or
the nuclear motion in molecules (femtoseconds, 1fs=10-15s). An
important goal for our future research will be to combine this ultrahigh
resolution in time with high spatial resolution at the intrinsic length scale
(1 Angstøm) of matter.
Contents:
1. Nuclear dynamics of molecules
in intense IR and XUV electric fields
1.1 Controlling the nuclear and electronic motion in molecules with short
light pulses
1.1.1
Control schemes for manipulating the shape of nuclear vibrational wave packets
1.1.2 Dissociative ionization of H2
in an attosecond pulse train and delayed laser pulse
1.1.3
Electron localization in
molecular fragmentation of H2 with CEP stabilized laser pulses
1.1.4
Laser control of the electronic motion and localization in H2+
in phase space
1.2 Nuclear wave-packet
dephasing and revivals in D2+
1.3 Towards the complete imaging of molecular dynamics with laser and XUV
pulses
1.3.1 Quantum-beat imaging of the
vibrational nuclear dynamics in diatomic molecules
1.3.2
Quantum-beat imaging of the rotational-vibrational nuclear dynamics in diatomic
molecules
1.4 Close-coupling calculations for H2
1.5 Moving to larger molecules:
Time-resolved fragmentation dynamics in N2, O2, and CO
2. Time-resolved
electronic dynamics in atoms and complex systems
2.1 Attosecond time-resolved
photoelectron spectroscopy of atoms
2.1.1 Coulomb-laser
coupling effects in attosecond time-resolved photoelectron spectra
2.1.2 Streaking and Wigner time delays in photoemission from atoms
2.1.3 Attosecond time-resolved
probing of instantaneous AC Stark shifts in helium atoms
2.1.4 Electron interference in atomic photoionization by a
single few-cycle IR laser pulse
2.1.5 Time-resolved autoionization
2.2 Time-resolved electronic dynamics in
complex systems
2.2.1 Attosecond time-resolved
photoelectron spectroscopy of metal surfaces
2.2.2 Laser-assisted photoemission from adsorbate-covered
metal surfaces:
Time-resolved
core-hole relaxation dynamics from sideband profiles
2.2.3 Dynamical image-charge effects in streaked photoelectron
spectra of metal surfaces
2.3 Electronic structure of flat and vicinal
surfaces
2.4 Image-potential states of single- and multi-walled carbon nanotubes
3. Laser-assisted collisions
4. Highly correlated
negative-ion resonances in photodetachment and electron scattering processes
1. Nuclear dynamics of molecules in intense
IR and XUV electric fields
1.1 Controlling the nuclear and electronic motion in molecules with short
light pulses
Project scope: To
develop numerical and analytical tools to efficiently predict the effects of
strong laser fields on the bound and free electronic and nuclear dynamics in
small molecules. Furthermore, we seek
to understand the degree to which the electronic motion in small molecules can
be steered by laser pulses, e.g., leading to the localization of an electron
near a given nucleus.
1.1.1 Control schemes for manipulating the shape of nuclear vibrational
wave packets
Recent
progress: We investigated the dissociation and ionization of D2 and D2+
in short intense laser pulses by applying wave-packet propagation methods. In
particular, we examined the possibility of manipulating the vibrational-state
decomposition of bound vibrational wave packets with a sequence of short
control laser pulses at minimal dissociative loss (Fig. 1).
|
|
Fig. 1. Ionization of D2 (ν=0) by a pump pulse, followed by the
modification of the vibrational wave
packet on the D2+ 1sσg+ potential curve by a control
pulse, and the final destructive analysis via Coulomb explosion imaging by
a probe pulse. |
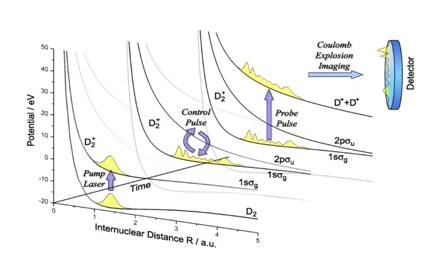
Ionization
of neutral D2 molecules by a short and intense pump laser pulse may
create a vibrational wave packet on the lowest (1sσg+)
adiabatic potential curve of the D2+ molecular ion [1,2].
We showed numerically that a single ultra–short intense near-infrared (800 nm)
control pulse with an appropriate time delay can strongly quench the
vibrational-state distribution of the nuclear wave packet by increasing the
contribution of selected stationary vibrational states of D2+ to
more than 50% [3]. We found that a second identical control pulse with a
carefully adjusted delay can further squeeze the vibrational state
distribution, likely without dissociating the molecular ion, thereby suggesting
a multi–pulse control protocol for generating (almost) stationary excited
nuclear wave functions (Fig. 2). The quality of this Raman–control mechanism
can be tested experimentally by Coulomb-explosion imaging, i.e., by fragmenting
the molecular ion with a probe pulse and by identifying the nodal structure of
the surviving vibrational state in the kinetic-energy-release (KER) spectrum of
the molecular fragments [1-4].
|
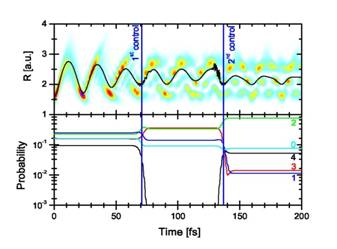
|
Fig. 2. Top: Time evolution of the nuclear wave function
probability density as a function of the internuclear distance R for two 6
fs, 1014 W/cm2 control pulses with delay times of τ1=70.7 fs and τ2=136.8 fs relative to the launch time (t=0).
The superimposed curve shows the expectation value <R>. Bottom:
Time evolution of a few stationary-vibrational-state contributions to the
wave packet. |
We compared results for our H2+ model
calculations that only include the lowest two coupled adiabatic potential
curves [3] with new full 3D calculations [5] and examined the possibility of manipulating the
vibrational-state decomposition of bound vibrational wave packets with a
sequence of up to eight control laser pulses at minimal dissociative loss. We find that
the precise timing between pump and control-laser pulses allows the direct
manipulation of the final vibrational state composition and dissociation
dynamics of the molecular ion. We showed that a significant enhancement of the
occupation of particular vibrational stationary state contributions can be
achieved for laser intensities below the onset of strong ionization.
|
|
Fig. 3. Stationary vibrational-state
composition of an initial Franck-Condon nuclear wave packet in D2+ after exposure
to a 6 fs 0.1 PW/cm2 Gaussian control pulse as a function of the
control-pulse delay. |
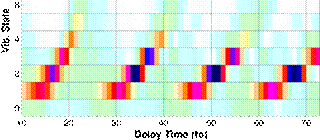
We found that a sequence of short control pulses can
effectively steer the nuclear motion in D2+ molecular ions and,
depending on the control-pulse delays, even stop a moving nuclear wave packet
to produce an excited stationary vibrational state (Fig. 3).
Future plans: Control schemes for quenching moving ro-vibrational
wave packets into stationary states using a sequence of control pulses will be
further examined and adjusted to model experimental conditions as closely as
possible.
Collaborators: Thomas Niederhausen, Fernando Martin (University of
Madrid, Spain)
[1] B. Feuerstein and U. Thumm, Phys. Rev. A 67, 043405 (2003).
[2] B. Feuerstein and U.
Thumm, Phys. Rev. A 67, 063408 (2003).
[3]
T. Niederhausen and U. Thumm, Phys. Rev. A 77, 013407 (2008).
[4] B. Feuerstein and U. Thumm, J. Phys. B 36, 707 (2003).
[5] T. Niederhausen, U. Thumm, and F. Martin, submitted
to Phys. Rev. A.
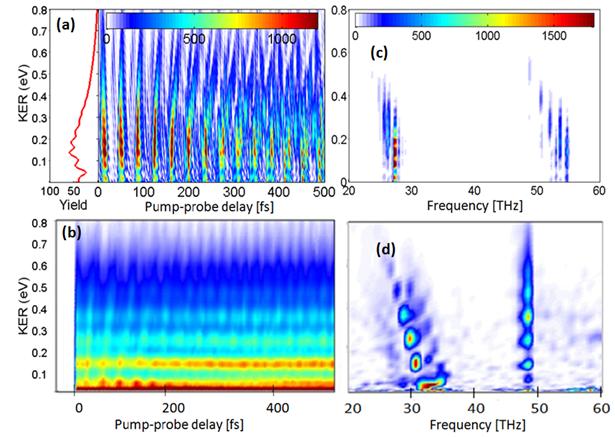
1.1.2 Dissociative ionization of H2
in an attosecond pulse train and delayed laser pulse
Recent progress: The ionization of H2 in a single
attosecond XUV pulse (SAP) generates a nuclear wave packet in H2+
which is entangled with the emitted photoelectron wave packet. The nuclear
wave-packet dynamics can be observed by dissociating H2+
in a delayed IR laser pulse. If H2+ is ionized by a
sequence of XUV pulses of an attosecond pulse train (APT) (Fig. 1), whether or
not the corresponding sequence of nuclear wave packets in H2+
is detected as a coherent or incoherent superposition depends on whether and
how the photoelectrons are observed. We simulated the nuclear dynamics in this
XUV pump - IR probe scenario and analyzed our numerical results for both,
single attosecond pump pulses and pump-pulse trains of different lengths and
temporal spacings between individual XUV pulses. By superimposing nuclear wave
packets in H2+ generated by individual pulses in the
pump-pulse train incoherently, we
calculated proton kinetic energy release (KER) spectra [1] in good qualitative
agreement with the experiment in reference [2].
|
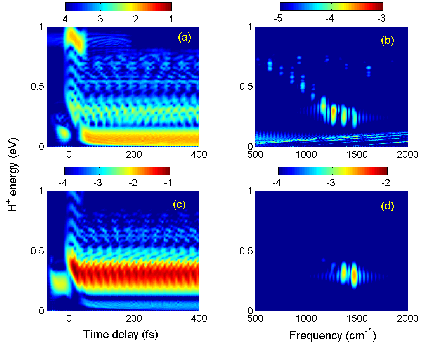
|
Fig. 1. (a) Schematic of the APT and IR laser field. The time delay Δt
is the offset between the centers of the APT and IR pulses. The pulse
duration of the IR pulse is τIR, and the separation between
subsequent attosecond pulses is Δt. (b) Relevant Born-Oppenheimer
potential curves of H2 and H2+. A sequence
of nuclear vibrational wave packets is generated on the 1sσg
electronic ground state potential curve of H2+ by
repeated ionization of H2 in subsequent XUV pulses of the APT. |
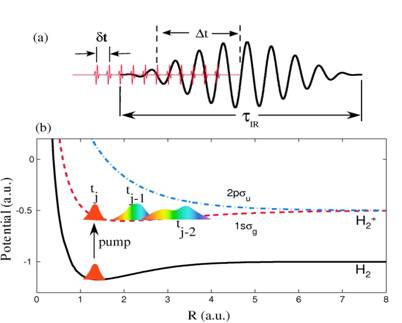
Since photoelectrons carry phase information, the
degree of coherence in the observed KER in an APT - delayed IR pulse experiment
will change if photoelectrons are detected in coincidence with molecular
fragments. Without the observation of coincident photoelectrons, we anticipate
the KER to be devoid of coherence effects between subsequently launched nuclear
wave packets, whereas coherence effects are expected to be most prominent if
photoelectrons are detected in extremely narrow momentum bins and coincident
with molecular fragments [1].
Future
plans: We
anticipate refined experiments in which protons and XUV-pulse-emitted electrons
are detected in coincidence. Assuming that such experiments can be carried out
with sufficiently large count rates, we predict an interesting transition from
an incoherent to a coherent superposition of nuclear wave packets by recording photoelectrons
in increasingly narrower momentum bins. For the coherent proton KER spectra, we
find an extremely sensitive dependence on the IR wavelength, that might be
exploited to characterize the IR laser pulse in terms of interference effects
(in both delay and proton energy) in fragment KER spectra (Fig. 2). With regard
to future numerical simulations, even for the simplest molecule, H2,
more work is needed in order to establish a firm lower limit for the effect of nuclear wave packet interferences on
KER spectra [1].
|
|
Fig. 2. Time-dependent proton energy distributions
(left column) and corresponding power spectra (right column) for H2
exposed to an APT and a delayed 30-cycle IR laser pulse with a peak intensity
of 1013 W/cm2. The APT consists of 14 alternating attosecond
XUV pulses. Maximal coherence is assumed for the superposition of individual
H2+ nuclear wave packets. (a) and (b): Results
for an IR carrier wavelength of λIR=800nm. (c) and (d):
Results for λIR =727nm. |
Collaborator: Feng He (SJTU Shanghai, PRC)
[1] F.
He and U. Thumm, Phys. Rev. A 81, 053413 (2010).
[2] F. Kelkensberg et al., Phys. Rev. Lett. 103, 123005 (2009).
1.1.3 Electron localization
in molecular fragmentation of H2 with CEP stabilized laser pulses
Recent
progress: Fully
differential data for H2 dissociation in ultrashort (6fs, 760nm),
linearly polarized, intense (0.44 PW/cm2) laser pulses with a
stabilized carrier-envelope-phase (CEP) were recorded with a reaction
microscope. Depending on the CEP, the molecular orientation, and the KER,
asymmetric proton emission at low KERs (0–3 eV) was measured [2] to be much
stronger than reported by previously [1]. Our wave packet propagation calculations
[2] reproduce the salient features and discard, together with the observed
KER-independent electron asymmetry, the first ionization step as the reason for
the asymmetric proton emission (Fig. 1).
Future
plans: Even though the
asymmetry in the experiment [2] shows a similar CEP and KER-dependence as in [3], the physical
situation considered there, an incoherent sum of vibrational states, is
different. Instead, for the experimental conditions in [2], a wave packet is
produced in the first step [4,5], pointing to the possible control of chemical
reactions through attosecond steering of electrons in a new type of
‘‘pump-control’’ experiment. Switching on the control laser at a time when the
wave packet approaches the (non-adiabatic) coupling region should strongly
enhance population transfer and asymmetry contrast. In this case control can be
achieved very efficiently by changing the pump-probe-delay, i.e., by guiding nuclear
wave packets through coupling regions where CEP stabilized pump-control schemes
steer the electronic motion on a sub-femtosecond time scale.
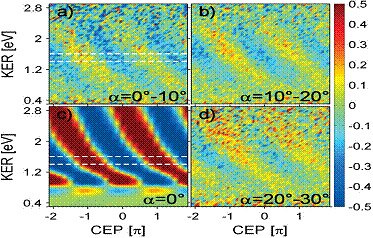
|
|
Fig. 1. Dissociation asymmetry in dependence
of the KER and the CEP for proton emission angles between (a) 0–10, (b) 10–20, and (d)
20–30 degrees with respect to the laser polarization axis. (c) Time-dependent Schrödinger
equation calculations. Since only relative CEPs were measured, the axes of
the experimental data were shifted to fit the calculation. |
Collaborators: Bernold Feuerstein and authors of ref. [2] (MPIK,
Heidelberg, Germany)
[1]
M. F. Kling et al., Science 312, 246 (2006).
[2] M. Kremer, B. Fischer, B. Feuerstein, V. L. B. De Jesus,
V. Sharma, C. Hofrichter, A. Rudenko, U.
Thumm, C. D.
Schröter, R. Moshammer, and J. Ullrich,
Phys. Rev. Lett. 103, 213003 (2009).
[3]
J. J. Hua and B. D. Esry, J. Phys. B 42, 085601 (2009).
[4] M. Winter, R. Schmidt, and U. Thumm, Phys. Rev.
A 80, 031401(R) (2009).
[5]
M. Winter, R. Schmidt, and U. Thumm, New J. of Phys. 12, 023023 (2010).
1.1.4 Laser control of the electronic motion and localization in H2+
in phase space
Recent
progress: The electronic dynamics in
a molecule driven by a strong laser field is complex and in part even
counterintuitive. As a prototype example, we have studied the electronic motion
inside dissociating H2+ molecules that are exposed to a
fs IR laser pulse [1]. The sensitive dependence of the correlated
electronic-nuclear motion can be explained in terms of the electronic momentum
distribution of the dissociating molecule. This distribution is dynamically
modulated by the nuclear motion and periodically shifted in the oscillating IR
electric field, leading to strong-field-modulated diffraction effects in the
correlated electronic-nuclear motion in dissociating molecular ions (Fig. 1).
Depending on the IR-laser intensity, the direction of the electronic motion can
follow or oppose the IR-laser electric force.
Our interpretation of this effect in terms of a Wigner
phase-space distribution [1] is based on the passage of electronic flux through
diffractive “momentum gates” of the two-center system that may or may not allow
the electron to transfer to the other nucleus (Fig. 2). It reveals that the
oscillating vector potential of the IR laser field periodically shifts these
gates, directing the electron through different gates at different laser
intensities. These results show how the internal electron dynamics in H2+
is driven by both the external laser field and diffraction effects.
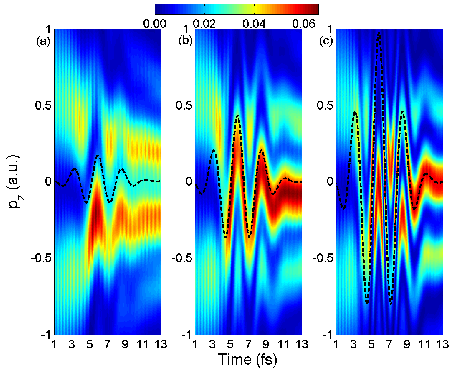
|
|
Fig. 1.
Electron
momentum distribution along the laser polarization during the dissociation of
H2+ in a 5.3 fs IR laser pulse with a time delay of 5.8
fs and intensities of (a) 3x1012,
(b)
2x1013, and (c) 1014
W/cm2. Dashed lines indicate the classical free-electron momentum
in the IR field, assuming zero initial momentum. The dissociating wave packet
was launched from the initial 1sσg onto the 2pσu state
of H2+ in a resonant single-photon transition, induced
by a 2-cycle, 106 nm, 1013 W/cm2 attosecond Gaussian
pump pulse. |
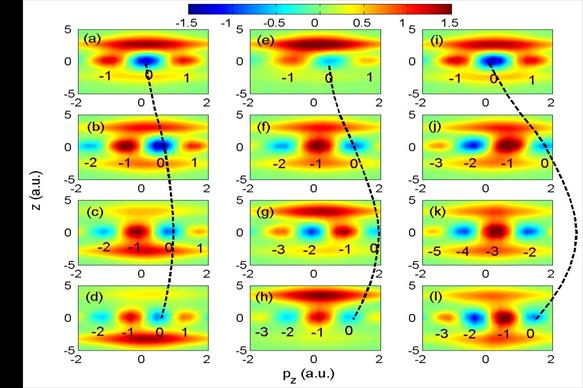
|
|
|
Fig. 2 Wigner distribution for IR laser intensities of 3x1012
(left), 2x1013 (middle) and 1014 W/cm2
(right column) and time delays of
4.5, 5.2, 5.8, and 6.5 fs (from the top to the bottom row). The dashed lines indicate
IR-laser-driven oscillations of the momentum gate that was initially centered at pz = 0. Future plans: We intend to further investigate the control - at a sub-fs time scale - of the
internuclear electronic dynamics in small molecules by tuning (IR) laser
parameters. Next, we plan to investigate the dynamics and control of
electronic motion at a sub-fs time scale in other molecules, including highly
symmetrical large molecules (C60). |
Collaborators: Feng He (KSU and SJTU Shanghai, PRC), Andreas Becker
(JILA, Boulder, CO)
[1] F. He, A. Becker, and U. Thumm, Phys. Rev. Lett. 101, 213002 (2008).
1.2 Nuclear wave-packet
dephasing and revivals in D2+
Project scope: To characterize
the laser-excited bound motion of coherent nuclear wave packets in small molecules in terms of decoherence,
dephasing, revivals, and competing dissociation, and to help with the interpretation of recent
experiments.
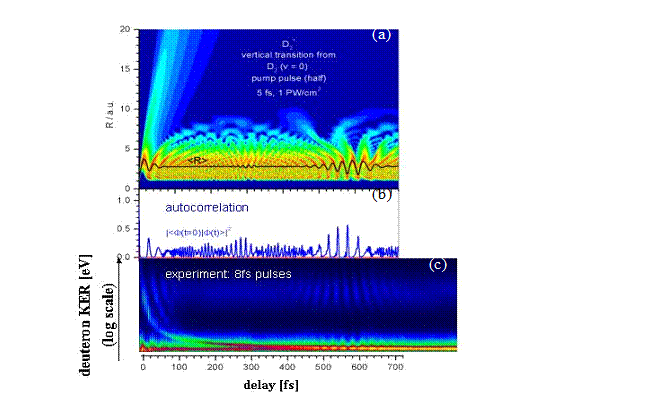 |
|
Figure
1a and b illustrates our reduced-dimensionality calculations [1,2] for the nuclear
wave-packet dynamics in D2+ following ionization of D2
(v = 0) in a 5 fs, 1015 W/cm2 pulse. After a few optical
cycles, the wave packet collapses due to the dephasing of its stationary
vibrational state components. The evolution of the nuclear probability density
(Fig. 1a) and autocorrelation function (Fig.
1b) shows revivals of the
wave packet 100 and 200 optical cycles, respectively, after the wave packet has
been launched. A second laser pulse, short and strong enough to ensure instantaneous
and complete ionization of D2+, can probe the time
evolution of the wave packet. Pump-probe experiments with 8 fs laser pulses on
D2 recorded the delay-dependent kinetic energy release of the
deuteron fragments [3,4] and reproduce the first half and full wave packet
revivals of our model calculation (Fig. 1c).
[1] B. Feuerstein and U. Thumm, Phys. Rev. A 67, 043405 (2003).
[2] B. Feuerstein and U.
Thumm, Phys. Rev. A 67, 063408 (2003).
1.3 Towards the complete imaging of molecular dynamics with laser and XUV
pulses
Project scope: We seek
to develop numerical and analytical tools to fully image the nuclear dynamics
in small molecules.
1.3.1
Quantum-beat imaging of the vibrational nuclear dynamics in diatomic molecules
Recent progress: In a proof-of-principles
effort, we introduced an internuclear-distance (R) -dependent harmonic imaging
technique that allows vibrational beat frequencies, molecular potential curves,
and the nodal structure of nuclear wave functions to be derived from measured
kinetic-energy-release (KER) spectra. In this method, the time-resolved KER
spectra of vibrating and dissociating D2+ molecules are
studied in comparison with the R-dependent harmonic analysis of the corresponding
wave packets [1, 2].
Our
calculations demonstrate that the obtained two-dimensional R-dependent
frequency spectra enable the characterization of the wave-packet dynamics and
directly visualize the field-modified molecular potential curves in intense,
ultra-short laser pulses [1], including “bond softening” and “bond hardening”
processes [3]. Figure 1 shows examples of this imaging scheme for the complete
mapping of molecular potential curves for the fundamental deuterium molecular
ion, for laser-free propagation of D2+ nuclear wave
packets (Fig. 1a) and including the interaction with a laser electric field
(Fig. 1b). These examples show how the molecular potential and its bound
vibrational wave function are modified by the added laser field.
|
|
Fig. 1. Power spectrum |w(R,f)|2
as function of the quantum-beat frequency f and R for a sampling time of T=3
ps. (a) Numerical field-free wave packet propagation of an initial Franck-Condon
distribution. (b) As (a) but with a 50 fs pedestal of 0.01 PW/cm2
preceding the probe pulse causing “bond softening” (BS) and “bond hardening”
(BH). (c) Experimental distribution extracted from coincident D+
pairs with vibrational (VIB) and rotational (ROT) contributions. White
contours: numerical results using the actual laser pulse profile shown in the
inset. |
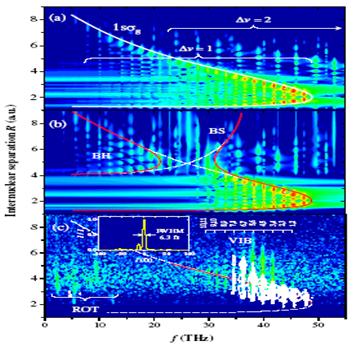
Our
method relies on the Fourier transformation, w(R,f), over a sampling time T of
the time- and R-dependent probability density w(R,t) of the D2+
nuclear wave packet. Applied to numerically propagated D2+
vibrational wave packets, it allows us to simulate the outcome of novel
experiments. The simulated experiments are assumed to be based on the
Coulomb-explosion mapping of pump-probe-delay (τ)-dependent KER spectra
and subsequent R-dependent harmonic analysis for finite T (0 < τ <
T). First experimental results [2], shown in Fig. 1c, reproduce the known
vibrational beat frequencies f and retrace the outer part of the potential
well. So far, the inner part of the potential well could not be observed due to
suppressed ionization rates at small R.
[1] U. Thumm,
T. Niederhausen, and B. Feuerstein, Phys. Rev. A 77, 063401
(2008).
[2] B. Feuerstein, T. Ergler, A. Rudenko, K.
Zrost, C.D. Schröder, R. Moshammer, J. Ullrich, T. Niederhausen, and U. Thumm,
Phys. Rev. Lett. 99, 153002
(2007).
[3]
M. Magrakvelidze, F. He, T.
Niederhausen, I. V. Litvinyuk, and U. Thumm, Phys. Rev. A 79, 033410 (2009).
1.3.2 Quantum-beat imaging of the
rotational-vibrational nuclear dynamics in diatomic molecules
Recent
progress: We investigated the extent
to which measured time-dependent fragment KER spectra and calculated nuclear probability
densities can reveal i) transition frequencies between stationary vibrational
states, ii) stationary rotational states and
ro-vibrational (RV) couplings, iii) the nodal structure of stationary
rotational and vibrational states, iv) field-free and laser-field-dressed
adiabatic electronic potential curves of the molecular ion, and v) the
progression of decoherence induced by random interactions with the environment
[1-4].
By
solving the TDSE in full dimensionality, we simulated the coherent evolution of
ro-vibrational nuclear wave packets and discussed their ro-vibrational dynamics
in D2+ [4,5] (Fig.
1) . Our imaging method is based on the Fourier transformation, w(R, θ,
f), over finite sampling times T, of the time-, internuclear distance (R)-, and
molecular orientation (θ)- dependent probability density w(R, θ, t)
of nuclear wave packet [3,4]. Our numerical results for ro-vibrational wave
packets demonstrate that the obtained two-dimensional R-dependent power spectra
enable the comprehensive characterization of the wave-packet dynamics and
directly visualize the laser-modified molecular potential curves in intense,
including `bond softening' and `bond hardening'
processes [1,4,5] (Fig.s 2 and 3). This harmonic-time-series analysis also
leads to a general scheme for the full reconstruction, up to an overall phase,
of the initial wave packet based on measured KER spectra [3].
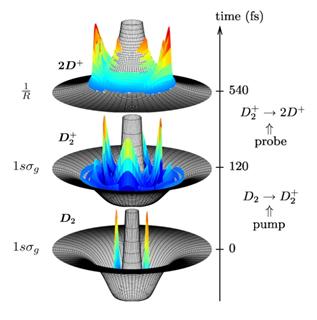
|
|
Fig. 1. Snapshots of the calculated time evolution of a ro-vibtational
nuclear wave packet in D20,+,2+. Bottom: At t=0 a pump laser pulse
ionizes D2 and excites the initial RV wave packet from the
1sσg, ν=0 state in D2 (bottom graph) to the
1sσg state of D2+ where it evolves,
continuously changing its distribution in
R and θ. Middle: Probability density of the wave packet in D2+
at t=120 fs. Ionization by a probe laser pulse after a delay of, e.g.,
τ=540 fs projects the wave packet onto the repulsive potential surface
of the 2D+-system (top graph), leading to fragmentation by CE. Top: Measurement of the KER of the D+ fragments as a function
of τ enables the characterization of the wave packet dynamics in terms
of R- and θ-dependent spectra. |
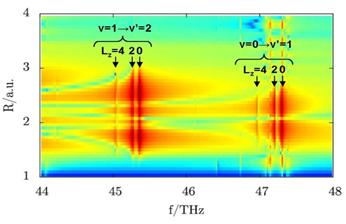
Including rotation of the molecular ion
(Fig. 2) [4,5], quantum-beat frequencies that correspond to a vibrational
transition ν→ν’ are split into multiple lines due to
rotational-vibrational coupling. These lines represent individual
angular-momentum contributions to the ro-vibrational wave packet (Fig. 2).
Based on numerical examples for the nuclear dynamics
without and under the influence of pulsed and continuum-wave (cw) laser light,
we discussed and quantified i) the signature of RV couplings in quantum-beat
spectra and ii) to what extent the quantum-beat analysis of measured
time-dependent fragment kinetic energy release spectra is expected to image the
laser-dressed RV structure of D2+ (Fig. 3).
|
|
Fig.
2. Angle-integrated
power spectra A(R,f) = |∫dθ w(R,θ,f)|2 for D2+
as a function of the beat frequency f and internuclear distance R. Due to
ro-vibrational couplings, lines for the same vibrational transition and
different angular momenta Lz do not coincide. Vibrational
transitions at larger Lz appear at lower frequencies. |
Future
plans: We intend to simulate the
extent to which the quantum-beat analysis of measured time-dependent fragment
KER spectra can quantify the laser-modulated ro-vibrational structure of H2+
and other diatomic molecules. Extending
this technique to more complicated polyatomic molecular systems and reaction
complexes may enable the investigation of molecular dynamics across the
(field-modified) potential barrier along a particular reaction coordinate, and,
thus, provide a basis for novel multidimensional optical-control schemes for
chemical reactions. We also envision to apply this method to quantify the
progression of decoherence in the nuclear motion based on a time series of KER spectra.
|
|||
|
Fig. 3. (a,c) Angle-integrated power
spectrum A(R,f). (e)
Internuclear-distance-integrated power spectrum W(θ, f) =
|∫dR w(R,θ,f)|2.
(b,d,f) Corresponding spectral
line intensities for the evolution of (a,b)
aligned and (c-f) rotating D2+ molecular
ions in a 1013 W/cm2 cw laser field. |
Collaborators: Rüdiger Schmidt, Martin Winter (Technical University
Dresden, Germany)
[1] M. Magrakvelidze, F. He, T. Niederhausen, I. V. Litvinyuk, and U. Thumm,
Phys. Rev. A 79, 033410 (2009).
[2] B. Feuerstein, T. Ergler, A. Rudenko, K. Zrost,
C.D. Schroeder, R. Moshammer, J. Ullrich, T. Niederhausen, and U. Thumm, Phys.
Rev. Lett. 99, 153002 (2007).
[3] U. Thumm,
T. Niederhausen, and B. Feuerstein, Phys. Rev. A 77, 063401
(2008).
[4] M. Winter, R. Schmidt, and U. Thumm, Phys. Rev.
A 80, 031401(R) (2009).
[5]
M. Winter, R. Schmidt, and U. Thumm, New J. of Phys. 12, 023023 (2010).
1.4 Close-coupling calculations for H2
Project scope: To investigate the simultaneous vibrational excitation and ionization
of H2 in a strong laser pulse by means of close-coupling
calculations.
Outline: We are in the process of
modeling the interaction of neutral H2 with strong few-cycle IR
laser pulses within a close-coupling calculation that retains the adiabatic
Born-Oppenheimer potential energy curves Vi (R), i=1...3, for the
electronic ground state of H2 and the ground and first exited states
of H2+. We plan to solve the time-dependent Schrödinger
equation, including all off-diagonal electric dipole couplings μij,
i,j=1...3, between the adiabatic electronic states. This approach describes the
coupled propagation of vibrational wave packets in H2 and H2+.
The wave packets in H2+ are due to the ionization of H2
while complementary wave packets in H2 are generated by “hole
burning” [1]. The process of hole burning is due to the predominant ionization
of H2 at larger internuclear distances that transforms the initial
stationary vibrational ground state of H2 into an outward moving
wave packet.
These close-coupling calculations are incomplete in
the sense that they do not resolve the motion of the ionized electron(s) and
thus require additional model assumptions for the dipole couplings. We intend
to investigate the prospect for conducting– at reasonable numerical expense – improved
close-coupling calculations that include electronic and nuclear degrees of
freedom. To guide us through these technically complex investigations of the
dissociative (single and double) ionization of H2, we view single-
and double ionization of the laser-plus-molecule system as Feshbach resonances
that can be parameterized in terms of Fano-resonance parameters. Numerical
applications will start with a limited basis consisting of the two lowest
Born-Oppenheimer electronic states of the molecular ion. We plan to proceed by
first modeling the bound-continuum couplings in terms of appropriate
parameters, and to continue with explicit calculations of the coupling matrix
elements. A serious challenge in the last step will be the adequate
representation of the electronic continuum in terms of discretized continuum
wave functions. Here, we will first explore the use of Weyl wave packets [2].
Our next goal is to repeat this sequence of steps for neutral molecules,
including two electronic continua and three electronic potential curves, adding
the ground-state potential curve of the neutral molecule.
[1] T. Niederhausen and U. Thumm, Phys. Rev. A 77, 013407 (2008).
[2] B. Bahrim und U. Thumm, Surf. Sci. 521, 84 (2002).
1.5 Moving to larger molecules: Time-resolved fragmentation dynamics in N2,
O2, and CO
Project scope: To develop
analytical and numerical tools to efficiently predict the effects of strong
laser and XUV fields on the bound and dissociative nuclear dynamics in heavy
diatomic molecules.
Recent
progress: We investigated the nuclear dynamics of electronically and vibrationally
excited heavy diatomic molecular ions by
applying intense ultrashort IR probe pulses and measuring the KER spectra as a
function of the pump-probe delay [1-4]. To analyze these spectra, we performed
wave-packet-propagation calculations on adiabatic molecular potential curves (Fig. 1). First, to identify relevant transiently populated
electronic states of the molecular ions,
we modeled
the pump step in Franck-Condon approximation and calculated the time evolution
of initial vibrational wave packets separately for selected molecular
potential curves.
|
|
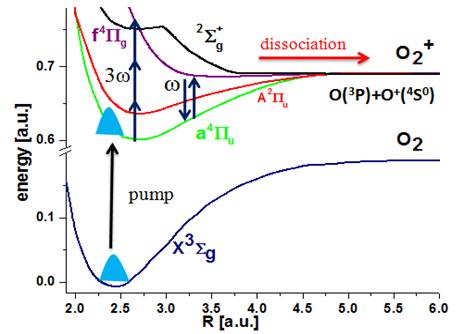
Fig. 1. Schematics for the
mapping of the nuclear dynamics in oxygen molecular ions. The pump-laser
pulse launches a nuclear wave packet onto the O2+
potential curves a4Пu and f 4Пg
by ionizing O2. After a variable time delay, an intense short
probe pulse can cause dissociation of the molecular ion through one or net
two photon processes. |
The comparison of calculated KER spectra as a
function of delay, quantum-beat frequency, and vibrational revival times for
one adiabatic curve at a time with experimental spectra served us as a guide
for selecting relevant electronic states of the
molecular ions. Next, we included probe-laser-induced dipole couplings between
the relevant molecular potential curves and compared the improved calculated
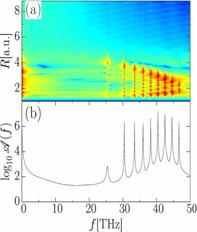
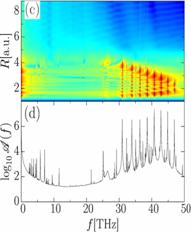
KER spectra with experimental data, in an attempt to reveal non-adiabatic effects
in measured KER spectra (Fig. 2). We employed the quantum chemistry code GAMESS
[5] to calculate molecular potential curve and dipole couplings between them
[4].
Future
plans: Measured
delay-dependent KER spectra of heavy diatomic molecules are difficults to
simulate theoretically and are not well understood. We believe that the
simultaneous study of measured and simulated KER spectra in both, time and
energy domains provides a powerful tool that we intend to refine in order to
disentangle the complicated ro-vibrational nuclear dynamics of laser-excited
(and ionized) molecules [4].
|
|
|
Fig. 2 (a,c) Calculated and (b,d)
measured [4] KER spectra for O2+ as a function of (a,b) pump-probe delay and (c,d)
quantum-beat frequency f. Calculated KER spectra include dipole-coupling of
the a 4Пu and f 4Пg
states by the 15 fs probe laser pulse with 3x1014 W/cm2peak
intensity. (c,d) Power spectra
obtained with a sampling time of 2 ps. |
Collaborators:
Maia Magrakvelidze, Lew Cocke, Itzik Ben-Itzhak (KSU), and
authors of ref.s [1-4]
[1] S. De, I. Bocharova, M. Magrakvelidze, D. Ray,
W. Cao, B. Bergues, U. Thumm, M. F. Kling, I. V.
Litvinyuk, and C. L. Cocke, Phys. Rev. A
82, 013408 (2010).
[2] I.A. Bocharova, A. S. Alnaser, U. Thumm, T.
Niederhausen, D. Ray, C.L. Cocke, and I.V. Litvinyuk, Phys. Rev. A 83, 013417 (2011).
[3]
M. Magrakvelidze, F. He, T. Niederhausen, I. V. Litvinyuk, and U. Thumm,
Phys. Rev. A 79, 033410 (2009).
[4] S. De, M.
Magrakvelidze, I. Bocharova, D. Ray, W. Cao, I. Znakovskaya, H. Li, Z. Wang, G.
Laurent, U. Thumm, M. F. Kling, I. V. Litvinyuk, I. Ben-Itzhak, and C. L.
Cocke, Phys. Rev. A, accepted for publication (Sept. 2011).
[5]
M. W.
Schmidt et al., J. Comput. Chem. 14, 1347 (1993).
2. Time-resolved electronic dynamics in atoms and complex
systems
We
model the
time-resolved IR-laser-assisted XUV photoelectron emission and Auger
decay in pump-probe-delay-dependent streaking experiments with atoms and
complex targets, such as clusters, carbon nanotubes, and surfaces.
2.1 Attosecond time-resolved
photoelectron spectroscopy of atoms
2.1.1 Coulomb-laser-coupling effects in
attosecond time-resolved photoelectron spectra
Project
scope: To
quantify and understand the effect of the
Coulomb interaction between the photoelectron and residual ion on the
photoemission dynamics and photoemission time delay.
Recent
progress: Photoionization by attosecond
XUV pulses into the laser-dressed continuum of the ionized atom is commonly
approximated in strong-field approximation (SFA), i.e., by neglecting the
Coulomb interaction between the emitted photoelectron and the residual ion
[1,2,3]. By solving the time-dependent Schrödinger equation (TDSE), we
identified a temporal shift δτ in streaked photoemission spectra that
is due to the Coulomb-laser coupling in the final-state and exceeds 50 as at
small photoelectron kinetic energies (Fig. 1). We expect the examination of
this shift to enable (i) the experimental scrutiny of effects that are due to
the combined action of Coulomb and laser forces on the photoelectron and (ii)
tests of theoretical approximations to the exact Coulomb-Volkov state of the photoelectron.
Within an eikonal (semiclassical) approximation, we derived an analytical
expression for this effect and assessed its accuracy in comparison with full
TDSE numerical results [4].
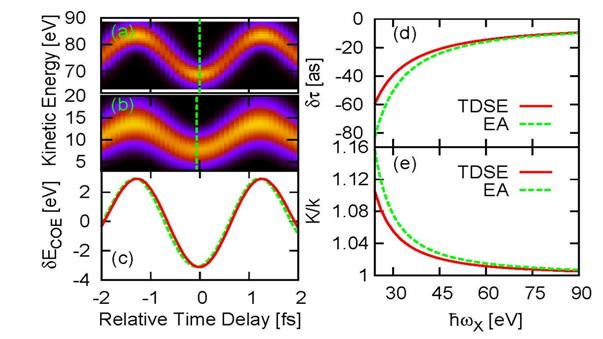
Fig. 1. Streaked photoemission from
1D model hydrogen atoms. TDSE calculations for XUV pulses with a central photon
energy of (a) ![]() 90 eV and (b) 25 eV. (c) Corresponding center-of-energy shifts δECOE(τ)
for
90 eV and (b) 25 eV. (c) Corresponding center-of-energy shifts δECOE(τ)
for ![]() =90 eV (solid line) and
25~eV (dashed line). To facilitate the identification of the relative temporal
shifts δτ, δECOE(τ,
=90 eV (solid line) and
25~eV (dashed line). To facilitate the identification of the relative temporal
shifts δτ, δECOE(τ, ![]() =90 eV) is normalized to the
=90 eV) is normalized to the
![]() =25 eV result. (d) δτ and (e) oscillation amplitude relative to
the SFA for TDSE (full line) and eikonal approximation (dashed line)
calculations.
=25 eV result. (d) δτ and (e) oscillation amplitude relative to
the SFA for TDSE (full line) and eikonal approximation (dashed line)
calculations.
Future
plans: We intend to (i) continue to
investigate the effect of interactions that are not included in SFA and (ii)
examine the influence of initial-state polarization in the streaking IR-laser
field on photoelectron spectra and time delays. We will seek contact with experimental
groups to explore the feasibility of and ideal parameters for the observation
of Coulomb-laser effects beyond the standard SFA in streaked photoemission
spectra [4].
Collaborator: Chang-hua Zhang (KSU)
[1] C.-H.
Zhang and U. Thumm, Phys. Rev. Lett. 102,
123601 (2009).
[2] C.-H. Zhang and U. Thumm, invited paper, XXVI
ICPEAC, Kalamazoo, Journal of Physics:
Conf. Series 194, 012055 (2009).
[3] C.-H. Zhang and U. Thumm, Phys. Rev. A 80, 032902
(2009).
[4]
C.-H. Zhang and U. Thumm, Phys. Rev. A 82,
043405 (2010).
2.1.2 Streaking and Wigner time delays
in photoemission from atoms
Project
scope: To compare different measures for the time
delay in photoemission from atoms by XUV photons and to examine Wigner and
streaking time delays [1-6] for the photoionization of atoms [3,5].
Recent
progress: Streaked photoemission
metrology allows the observation of an apparent relative time delay between the
detection of photoelectrons (Pes) from different initial electronic states [1-3,6,7].
Theoretically, photoemission delays can be defined based on (i) the phase shift
the photoelectron wavefunction accumulates during the release and propagation
of the PE (``Wigner delay") and, alternatively, (ii) the streaking trace
in the calculated photoemission spectrum (``streaking delay") , while experimentally time delays
can only be deduced from streaked PE spectra [1,3,6]. We
investigated the relation between Wigner and streaking delays in the
photoemission from atoms and solid surfaces. For surfaces and assuming a
vanishing IR-skin depth, both Wigner and streaking delays can be interpreted as
an average propagation time needed by photoelectrons to reach the surface,
while the two delays differ for non-vanishing skin depths [3,4]. For atomic
targets, the difference between Wigner and streaking delays depends on the
range of the ionic potential [3].
Future
plans: We intend to clarify the precise
interpretation of and relations between different time-delay measures based on
specific numerical examples for photoemission from atoms and surfaces.
Collaborator: Chang-hua Zhang (KSU)
[1] C.-H.
Zhang and U. Thumm, Phys. Rev. Lett. 102,
123601 (2009).
[2] C.-H. Zhang and U. Thumm, invited paper, XXVI
ICPEAC, Kalamazoo, Journal of Physics: Conf. Series 194, 012055 (2009).
[3] C.-H.
Zhang and U. Thumm, Phys. Rev. A 84,
033401 (2011).
[4] C.-H. Zhang and U. Thumm, Phys. Rev. A,
submitted; http://arxiv.org/abs/1102.0751
[5]
C.-H. Zhang and U. Thumm, Phys. Rev. A 82, 043405 (2010).
[6]
M. Schultze et al., Science 328, 1658 (2010).
[7]
A. L. Cavalieri et al., Nature 449, 1029 (2007).
2.1.3 Attosecond
time-resolved probing of instantaneous AC Stark shifts in helium atoms
Project scope: To model,
calculate, and understand the excitation and ionization of atoms in an IR-laser
field with sub-optical-cycle (TIR ) time
resolution.
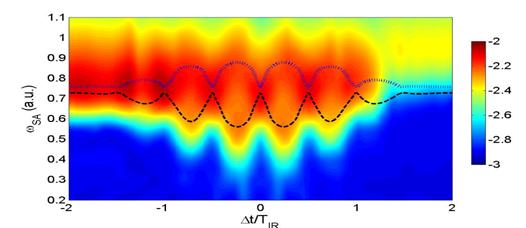
Recent
progress: The
role of laser-dressed highly excited energy levels in atomic excitation and
ionization has been studied recently using attosecond technology [1,2]. We
followed up on these studies and showed that this pump-probe technique also
enables the measurement of instantaneous
level shifts of bound atomic [3] (and molecular [4]) states in optical electric
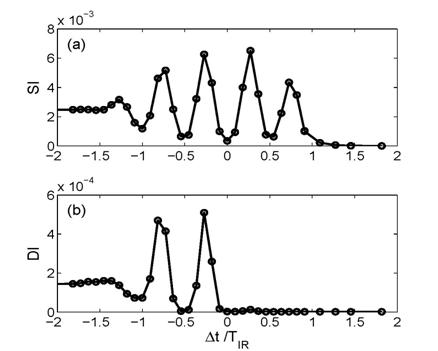
fields (Fig. 1). We demonstrated how the control of instantaneous level shifts
can be exploited to gate strong-field phenomena, such as non-sequential double
ionization (NSDI) [3] (Fig. 2).
Based on numerical
solutions of TDSE for either one or two active electrons, we developed a method
for observing time-dependent instantaneous level shifts in an oscillating
strong IR field, using a single tunable XUV pulse to probe excited states of
the perturbed atom. We assumed IR-laser fields with negligible distortion of the He ground
state, which are, however, strong enough to couple low-lying excited and
continuous states, inducing noticeable level splitting, shift, and decay. We
fixed the number of XUV cycles and varied the central frequency ωSA of the XUV pulse (Fig. 1). Key to our
investigation is the observation that, for a given ωSA of the
attosecond XUV pulse and depending on the delay ∆t between pump and probe
pulse, the IR pulse may shift low-lying bound states into or out of resonance
with one-photon excitations from the He ground state. The excited atom may then
be easily ionized by the remaining IR pulse. Applying the SA pulse while instantaneous
level energies are off (in) resonance with ωSA, results in less
(more) excitation and thus less (more) ionization out of excited states. This
suggests that detection of the ionization probability as a function of ωSA
and ∆t can be used to experimentally track instantaneous Stark shifts
[3,5].
|
|
|
|
Fig. 1. Ionization probabilities (logarithmic color scale) of He calculated in single-active-electron-approximation
as a function of the center frequency ωSA of the (single) attosecond (SA) XUV pulse and time delay Δt between the SA and IR laser
pulses in units of the IR laser period TIR.
Superimposed dashed and dotted curves show the quasi-static energy
differences between the Stark-shifted 1s
and 2s (dashed line) and 1s and 2p (dotted line) levels. |
|
|
|
Fig. 2. Probabilities for single ionization (SI) (a) and double ionization (DI) (b) as a function of the time delay between SA and IR pulses. The
SA pulse has a central frequency of 0:76 a.u. and a peak intensity of
2x1013 W/cm2. The IR pulse has a central wavelength of
800 nm and a peak intensity of 3x1014 W/cm2. The
simulation results (circles) are interpolated by lines. |
Future
plans: The proposed method (i) allows the detection of
instantaneous atomic energy gaps with sub-laser-cycle time resolution and (ii)
can be applied as an ultrafast gate for more complex processes such as NSDI [3].
We intend to continue to search for ideal laser parameters and
targets for the observation, with sub-IR-cycle resolution, of AC Stark shifts
in delay-dependent single and double
ionization probabilities. This may lead to new schemes for the coherent control
of NSDI, high harmonic generation, and molecular dissociation, for which we
hope to find suitable proof-of-principle examples.
Collaborators: Feng He (SJTU Shanghai, China), Camilo Ruiz (CLPU
Salamanca, Spain), Andreas Becker (JILA, Boulder, CO)
[1]
P. Johnsson et al., Phys.Rev. Lett. 99, 233001 (2007).
[2]
P. Ranitovic et al., New J. Phys. 12, 013008 (2010).
[3] F. He, C. Ruiz,
A. Becker, and U. Thumm, Phys. Rev. Lett, submitted; arxiv.org/abs/1105.5204
[4] F. He, A. Becker, and U. Thumm, Phys. Rev. Lett. 101, 213002 (2008).
[5]
H. Wang, M. Chini, S. Chen, C.-H. Zhang, F. He, Y. Cheng, Y. Wu, U. Thumm, and
Z. Chang, Phys. Rev. Lett. 105, 143002 (2010).
2.1.4 Electron interference in
atomic photoionization by a single few-cycle IR laser pulse
Project scope: To model,
calculate, and understand interference structures in photoelectron spectra, in
particular, to distinguish structures that are due to above-thresholds
ionization from intra-IR cycle electron interferences.
Recent
progress: We analyzed recently measured [1] interference
patterns in momentum-resolved single-ionization photoelectron spectra from He
targets with regard to the interference of specific contributions to calculated
photoelectron spectra that originate from a few selected sub-laser-cycle
time intervals during the laser-atom interaction (Fig. 1). For contributions
from just two such narrow time intervals that are centered at successive maxima
of the laser-electric field with lengths of a few attoseconds, our calculations
reproduce some of the measured interference structure in the momentum-resolved
spectra. By selecting photoelectron wave packets that are released with inter-
or intra-IR-cycle spacings [2], we were able to distinguish known
above-threshold-ionization (ATI) interferences and non-ATI interference structures
in simulated photoelectron spectra [3].
|
|
Fig. 1. IR-laser electric field (solid line) and field envelope (dotted
line). The yellow/gray shaded area shows the photoelectron yields calculated
in strong-field approximation (in arbitrary units). Also shown are two
classical trajectories (dashed lines) for electrons that are emitted within
an IR cycle, at times t1 and t2 and detected with
momenta pt1 and pt2, respectively. The oscillation
amplitudes of these trajectories are given in arbitrary units. |
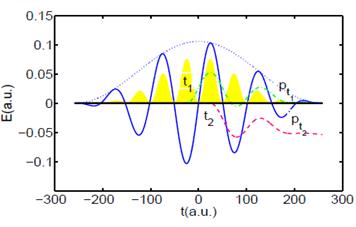
Future
plans: We
intend to provide a more complete interpretation of interferences in the
momentum-resolved photoionization of atoms by single few-cycle IR pulses in
terms of a semiclassical analysis of relevant electron trajectories.
Collaborator: Aihua Liu (KSU)
[1] R. Gopal et al., Phy. Rev.
Lett. 103, 053001 (2009).
[2] D. G. Arbo et al.,
Phys. Rev. A 81, 021403(R) (2010).
[2] “Sub-optical-cycle
photoelectron-wave-packet interference in few-cycle laser pulses”, A.
Liu and U. Thumm, in preparation.
2.1.5 Time-resolved autoionization
Project
scope: To simulate absorption spectra for the
time-dependent autoionization from atoms.
Recent
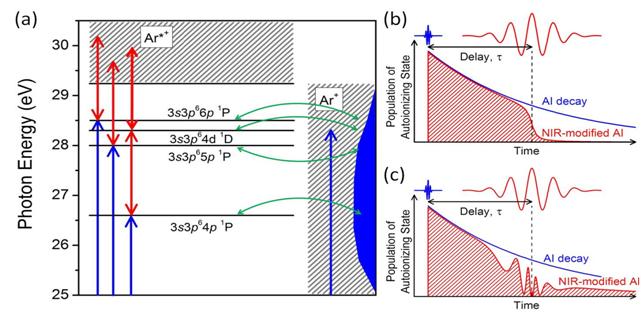
progress: Autoionization in argon
atoms was recently studied experimentally by transient absorption spectroscopy
with isolated attosecond pulses [1] (Fig. 1). The peak position, intensity,
line width, and shape of the 3s3p6np 1P Fano resonance series (26.6-29.2 eV) were modified
by intense few-cycle near infrared laser pulses, while the
delay between the attosecond pulse and the laser pulse was changed by a few
femtoseconds. Our numerical simulations [1] revealed that the
experimentally observed splitting of the 3s3p64p 1P line is caused by the coupling between two short-lived highly-excited
states in the strong laser field (Fig. 2).
Future
plans: Our simulations [1] of the laser-induced coupling of the 3s3p64p and 3s3p64d autoionizing states were performed
based on the heuristic interaction matrix elements [2]. In our model, the 3s3p65p and 3s3p66p states as well as the Ar*+
(3s3p6εl) continuum
are ignored, but coupling to the Ar+ (3s23p5εl) continuum via configuration interaction
is included. We plan to develop a fully ab-initio time-dependent calculation
for He atoms and use this calculation to scrutinize our semi-heuristic
calculation in Ref. [1].
|
|
|
Fig. 1. (a) Schematic representation of argon autoionizing states exposed
to the strong laser field. The blue arrows indicate the attosecond XUV
excitation of the ground state to the 3s3p6np
1P states as well as to the Ar+ (3s23p5εl)
continuum. The red arrows indicate the NIR laser coupling between the
autoionizing states and the Ar*+ (3s3p6εl) continuum
or to 3s3p6nl
autoionizing states. The configuration interaction (green arrows) couples all
autoionizing states to the Ar+ continuum. (b) Autoionization decay modified by NIR laser-induced coupling
to the Ar*+ (3s3p6εl) continuum. Ionization truncates the autoionization
decay, resulting in a shorter lifetime and a broader, shifted resonance peak.
(c) Autoionization decay modified
by NIR laser-induced coupling to 3s3p6nl
autoionizing states. Rabi oscillation between the two states results in AC
Stark-like splitting. |
|
|
|
Fig. 2. Simulated dipole radiation
spectrum of laser-induced coupling of the 3s3p64p
and 3s3p64d autoionizing
states. The XUV laser has a pulse duration of 140 as and intensity of 1010
W/cm2. The NIR laser had a pulse duration of 8 fs and intensity of
(a) 5×1011 W/cm2
and (b) 1×1012 W/cm2. |
Collaborators: Authors of ref. [1] (KSU and University of Central
Florida, Orlando)
[1]
H. Wang, M. Chini, S. Chen, C.-H. Zhang,
F. He, Y. Cheng, Y. Wu, U. Thumm, and Z. Chang, Phys. Rev. Lett. 105, 143002 (2010).
[2] C.-H.
Zhang and U. Thumm, Phys. Rev. A 80, 032902 (2009).
2.2 Time-resolved
electronic dynamics in complex systems
2.2.1 Attosecond
time-resolved photoelectron spectroscopy of metal surfaces
Project scope: To model the
time-resolved photoelectron (PE) emission in pump-probe and streaking
experiments with complex targets.
Recent
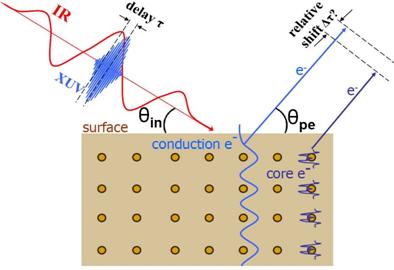
progress: In attosecond time-resolved
PE streaking experiments on metal surfaces, attosecond extreme ultraviolet (XUV)
light pulse are used to release electrons from either bound core levels or
delocalized conduction-band states (Fig. 1). The released electrons get exposed
to (“streaked by”) the same IR probe pulse, that was also used to generate the
XUV pulse via harmonic generation. The two laser pulses are thus synchronized
with a precisely adjustable time delay τ, and the measured asymptotic PE
kinetic energy E depends on τ. By varying τ, the time-resolved PE
kinetic energy distribution P(E,τ) can be recorded. This method was first
successfully applied to isolated atoms in the gas phase [1] and, more recently,
to tungsten [2] and platinum [3] surfaces. By using attosecond streaking spectroscopy,
Cavalieri et al. [2] measured a
relative delay of 110 ± 70 as between the detection of electrons that are photoemitted
by absorption of a single XUV photon from 4f-core
and conduction-band levels. Due to their different initial energies, PEs from core
and conduction-band levels can be easily separated in the energy-differential
spectra.
|
|
Fig. 1. Attosecond streaking
spectroscopy at metal surfaces. Delocalized conduction electrons and
localized core-level electrons are released by an attosecond XUV pulse
(photoelectric effect) and streaked by an IR laser pulse. By changing the
delay between the two pulses, the delay between the detection of photoelectrons
that originate in conduction band and core levels can be measured. |
For
the W (110) surface we use measured values for the work function (W=5.5 eV),
Fermi energy (EF = 4.5 eV), and lattice constant in direction perpendicular
to the surface (a = 3.13 Å). We calculated the energy-resolved spectra PCB
(E, τ) and P4f (E,
τ) for the two groups of PEs as a function of τ [4,5]. The comparison
of experiment and theory in Fig. 2 shows that our IR pulse modulation of the PE
kinetic energy agrees with the experiment. In order to find the temporal shift
between the two calculated spectra in the right column of Fig. 2, we calculated
their center-of-energies ECOE (τ). The temporal shift between 4f core-level and conduction-band
electrons is recognizable in Fig. 3 for both experimental data and calculation.
|
|
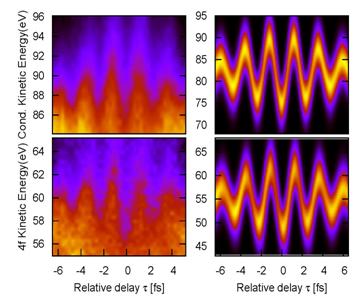
Fig. 2. Time-resolved photoelectron spectra for emission out of the
conduction band (top) and 4f core level (bottom) of a W (110) surface, as a function of the delay between
the XUV and IR pulses. Experimental results [2] (left) in comparison with our calculation [4] (right). |
|
|
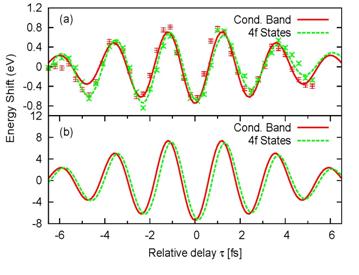
|
Fig. 3. Streaked electron spectra for photoemission from conduction-band and
4f core levels of a W (110) surface.
Center-of-energy shift as a function of the delay between the XUV
and IR pulse. (a) Experimental
results from [2]. The damped sinusoidal curves are fits to the raw
experimental data (points with error bars). (b) Calculated results showing a relative shift of 110 as between
the two groups of electrons. For better comparison, energies for the 4f photo-electrons are multiplied by a
factor 2.5 in (a) and 1.1 in (b). |
|
Future
plans: We
intend to refine our calculations in [4] by including diffraction effects
during the propagation of PEs inside the solid and by allowing for arbitrary
angles of IR-light/X-ray incidence and PE emission: (i) Since the transport (in our model the electron
mean-free path λ) depends on the PE kinetic energy, and thus the XUV
frequency ωX, we anticipate future tests of this predicted
sideband enhancement effect in experiments with tunable ωX. (ii)
While we believe fully localized states are a good approximation for the 4f state in tungsten [4,5], the fully
delocalized plane wave (jellium) approximation [6] does not take into account
that 5d6s conduction-band states in
tungsten have some localized character as well. For a fixed value of λ=5
a.u., allowing for partial localization of the conduction-band states is
expected to decrease the temporal shift between core and conduction-band
levels. In an improved model, this decrease could be compensated by increasing
λ to a value closer to accepted values for tungsten [5,7]. We thus plan to
improve our modeling of the metal conduction band and the propagation of
photoelectrons inside the solid.
Collaborator: Chang-hua Zhang (KSU)
[1] M.
Hentschel et al., Nature 414, 509 (2001); R. Kienberger et al., Nature 427, 817 (2004).
[2] A. L.
Cavalieri et al., Nature 449, 1029 (2007).
[3] L.
Miaja-Avila et al., Phys. Rev. Lett. 101, 046101 (2008).
[4] C.-H. Zhang and U. Thumm, Phys. Rev. Lett.
102, 123601 (2009).
[5] C.-H. Zhang and U. Thumm, invited paper, XXVI
ICPEAC, Kalamazoo, Journal of Physics: Conf. Series 194, 012055 (2009).
[6] A. Schmitz, J. Shaw, H. S. Chakraborty,
and U. Thumm, Phys. Rev. A 81,
042901 (2010).
[7] C.-H. Zhang and U. Thumm, Phys. Rev. A 80, 032902
(2009).
2.2.2 Laser-assisted
photoemission from adsorbate-covered metal surfaces:
Time-resolved core-hole relaxation
dynamics from sideband profiles
Project
scope: We attempt to model the
time-resolved photoelectron (PE) emission and Auger decay in pump-probe
experiments with adsorbate-covered surfaces and thin films.
|
|
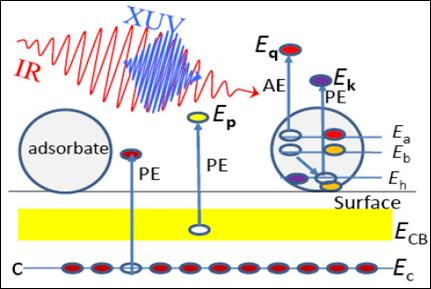
Fig.1. Sketch of the emission of
substrate (core level and conduction band) and adsorbate (core level and
Auger) electrons by an XUV pulse. Photoreleased electrons are exposed to a
weak delayed IR pulse. |
Recent
progress: Illumination of an adsorbate-covered metal
surface with an XUV pulse and a delayed IR laser pulse (Fig. 1) can result in
sidebands in the PE spectra [1]. We developed a theoretical model for the
delay-dependent photoemission process and showed how the relaxation dynamics of
XUV-induced core-level holes in adsorbate atoms can be deduced from the
temporal shift between sideband peaks in the spectra of secondary adsorbate
(Auger) electrons and conduction band (CB) PEs from the substrate (Fig. 2) [2-4].
|
|
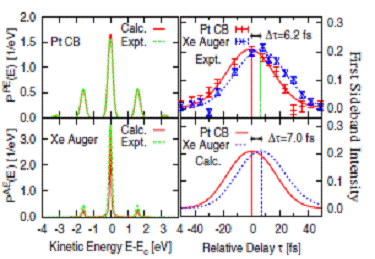
Fig. 2. Theoretical [4] and
experimental [1] PE spectra for laser-assisted photoemission from a
Xe/Pt(111) surface. Left: Sideband
intensities for no delay (τ=0) between XUV and IR pulses for XUV-emitted
conduction-band electrons (Pt CB) from the Fermi level (top) and Xe Auger electrons (bottom). Right: Experimental (top) and
calculated (bottom) first sideband intensities for Pt CB electrons and Xe Auger
electrons, revealing a temporal shift Δτ. Sideband intensities in
the AE spectra are multiplied by a factor of 2.16. |
In
comparison with gaseous targets, we found a characteristic sideband-intensity
enhancement in the laser-assisted photoemission from the substrate core-levels
[3,4]. This effect can be tested in experiments with tunable XUV wavelength.
Our calculated PE spectra support first time-resolved experiments for
Xe-covered Pt(111) surfaces, enabling the direct analysis in the time domain of
surface dynamical processes. This intensity redistribution between the main and
sideband peaks in core level photoelectron spectra from metals surfaces is
related to the transport of photoreleased electrons in the substrate [2,4].
Future
plans: We intend to systematically apply quantum mechanical S-matrix theory to
the study of time-resolved core-hole relaxation spectroscopy on adsorbate-covered
surfaces.
Collaborator: Chang-hua Zhang (KSU)
[1] L. Miaja et al., Phys. Rev. Lett. 101,
046101 (2008).
[2]
C.-H. Zhang and U. Thumm, Phys. Rev.
Lett. 102, 123601 (2009).
[3] C.-H. Zhang and U. Thumm,
invited paper, XXVI ICPEAC, Kalamazoo, Journal of Physics: Conf. Series 194,
012055 (2009).
[4] C.-H. Zhang and U. Thumm, Phys. Rev. A 80, 032902
(2009).
2.2.3 Dynamical
image-charge effects in streaked photoelectron spectra of metal surfaces
Project scope: To provide theoretical guidance towards the application of time-resolved
streaked photoelectron spectroscopy for the investigation of ultrafast
plasmonic dynamics in complex targets, such as clusters, carbon nanotubes, and
surfaces.
|
|
Fig. 1. A moving photoelectron
excites surface and bulk plasmons in the metal substrate that lead to a
dynamical redistribution of surface charge. This charge redistribution, in
turn, acts back on the photoelectron and changes the photoemission time
delay. In the limit where the photoelectron is far away from the surface and at
rest, the net effect of the charge redistribution amounts to the interaction between
the photoelectron and its fictitious image charge. |
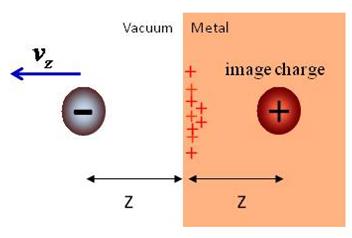
Recent
progress: The release of conduction-band electrons from a metal surface by a
sub-femtosecond XUV pulse, and their propagation through and near the solid
[1,2,3,4], provokes a dielectric response in the solid that acts back on the photoelectron
(PE) wave packet (Fig. 1). We modeled the response of the metal due to
excitation of bulk and surface plasmons induced by the creation and propagation
of PEs in the solid in terms of an effective potential that depends on the
velocity of the PE. We numerically calculated the (wake) potential associated
with this PE self-interaction and showed that it induces a considerable
XUV-frequency-dependent temporal shift in streaked-photoemission spectra [5],
suggesting the observation of the ultrafast solid-state dielectric response in
contemporary streaked photoemission experiments [4]. We are currently analyzing
the dependence of this relative shift on the XUV frequency as well as on
solid-state characteristics, such as the bulk-plasmon frequency, the IR-skin
depth, and the PE transport in the solid (Fig. 2).
Future
plans: We plan to (i) further improve our modeling
of the transport (including diffraction effects) of released PEs inside the
substrate and (ii) collaborate with experimental groups to explore the feasibility of and ideal
parameters for the observation of plasmon response effects ( i.e., the
time-resolved creation of “image charges”)
during and after the XUV-pulse-triggered release of PEs from metal
surfaces [4,5].
|
|
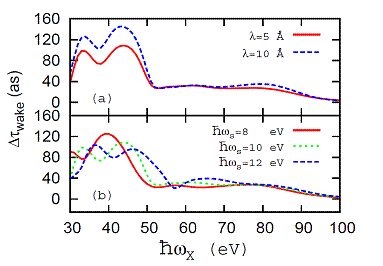
Fig. 2. Contribution Δτwake
to the streaking delay in photoemission from a metal surface due to the
dynamic plasmon response. The relative delay Δτwake is
shown as a function of the XUV frequency ωX for vanishing IR
skin depth and at different (a) photoelectron
mean-free paths λ and (b)
surface-plasmon frequencies ωS. We obtained Δτwake
as the difference of time delays in calculated streaked photoemission spectra
that included either the dynamical plasmon response or just the static image
potential. |
Collaborator: Chang-hua Zhang (KSU)
[1] C.-H.
Zhang and U. Thumm, Phys. Rev. Lett. 102,
123601 (2009).
[2] C.-H. Zhang and U. Thumm, invited paper, XXVI
ICPEAC, Kalamazoo, Journal of Physics: Conf. Series 194, 012055 (2009).
[3] C.-H. Zhang and U. Thumm, Phys. Rev. A 80, 032902
(2009).
[4]
A. L. Cavalieri et al., Nature 449, 1029 (2007).
[5] “Probing
dielectric response effects with attosecond time-resolved streaked
photoelectron spectroscopy of metal surfaces”, C.-H. Zhang and U. Thumm,
Phys. Rev. A., submitted; http://arxiv.org/abs/1102.0751
2.3 Electronic structure of flat and vicinal
surfaces
Project scope: (i) To develop models for the
efficient representation of the valence electronic structure of complex systems
in terms of single-electron effective potentials and (ii) to use these
effective potentials in modeling the interaction of particles and (XUV and IR)
radiation with metal surfaces and carbon nanotubes.
Recent
progress: We developed a new set of
computer programs to calculate the ground-state electronic structure of
arbitrarily shaped metallic surfaces and tested our codes in applications to
flat and vicinal metal and semi-conductor surfaces [1,2]. We used a density
functional model for the ground-state electronic structure of the surface that
included linear and quadratic electronic response terms and heuristic core
potentials centered at the lattice points, in order to provide realistic,
self-consistent surface potentials and corresponding electronic charge
densities (Fig. 1). We employed these potentials to model the charge-transfer
dynamics during ion-surface collisions, based on a Newns-Anderson approach and
including image-charge interactions and electron translation factors [3,4].
|
|
|
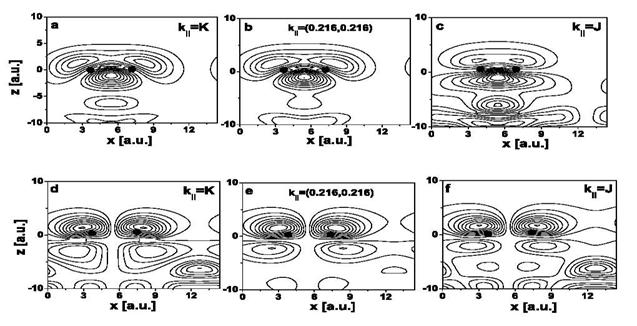
Fig. 1. Charge densities of (a)–(c)
π bonding and (d)–(f)
π* anti-bonding surface states on the (2 × 1)-reconstructed
Si (100) surface. The first 4.5 layers are shown. The parallel Bloch momentum
k|| changes along a straight-line connecting the
high-symmetry end points K and J in the surface Brillouin zone. The positions of
the Si cores forming the surface dimer are indicated by dots [4]. |
|
|
Fig. 2. Negative-ion survival as a function of the exit velocity
component normal to the surface for incident 1 keV hydrogen anions that are
reflected on a (2×1)-reconstructed
Si(100) surface. The angles of incidence (relative to the surface plane) are between
30 and 250. Upward pointing triangles give final anion
yields for projectiles moving perpendicularly to Si-dimer rows. Inverted
triangles correspond to trajectories oriented parallel to dimer rows. Circles
show the measured [5] negative-ion yields on Si surfaces. |
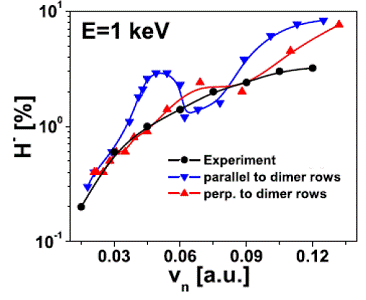
Employing
these effective potentials, we calculated the yield of outgoing negative
hydrogen ions after scattering off a reconstructed Si (100) surface [3,4]. We find
that the outgoing H- fraction is mainly determined by electron
capture from dangling-bond surface-state resonances at relatively large distances
from the surface. Our results are in fair agreement with the experimental
results of Maazouz et al. [5] and
with recent independent calculations by Garcia et al. [6] (Fig. 2).
Future plans: We will test our numerical implementation of this
method first by comparing workfunctions for flat surfaces and work function
changes due to vicinal superstructures with published data. We intend to investigate resonance formation, charge
exchange, and time-resolved photoemission near vicinal and other nano-structured
surfaces. In particular, we plan to assess the importance of lateral
confinement effects [7] (evidence of which was found in photoemission
experiments) on ion neutralization and photoemission.
Collaborator: Boyan Obreshkov (KSU)
[1] B. Obreshkov and U. Thumm, Phys. Rev. A 74,
012901 (2006).
[2]
B. Obreshkov and U. Thumm, Surf.
Sci. 601, 622 (2007).
[3]
B. Obreshkov and U. Thumm, Phys.
Rev. A 76, 052902 (2007).
[4] B. Obreshkov and U. Thumm, Phys. Rev. A 83, 062902 (2011).
[5] M. Maazouz et al., Surf. Sci. 398, 49 (1998).
[6] E. A. Garcia et al., Surf. Sci. 600,
2195 (2006).
[7] U. Thumm, P. Kürpick, and U. Wille, Phys. Rev. A 61, 3067 (2000).
2.4 Image-potential states of single- and multi-walled carbon nanotubes
Project scope: (i) To develop
models for the efficient representation of the valence electronic structure of carbon
nanotubes (CNT) in terms of single-electron effective potentials and (ii) to use these potential for the
investigation of two-photon photoemission processes.
Recent progress: We have investigated the formation of
image-potential states near the surfaces of single- and multi-walled CNT (Fig.
1). These states are confined between the self-induced image potential on the
vacuum side and the surface barrier. We calculated binding energies and wave
functions by modeling the interactions inside the nanotube with a cylindrical
jellium-like short-range potential that is parameterized to ensure the correct
vacuum to surface transition.
We found an interesting variation of the image-state
properties on the nanotube diameter [1], which is due to the difference in the
radial dependence of the induced image potential and the centrifugal potential.
Our results predicting the existence of image-potential states in CNTs were
confirmed in recent time-resolved photoemission experiments [2].
|
|
Fig. 1. Visualization of an image electron wave function
for a multi-walled carbon nanotube. |

Future plans: Image states are sensitive probes of the
dielectric response of CNTs. Their properties are affected by the anisotropic
polarizability of the tube. A realistic description of electronic image-state
spectra thus requires detailed knowledge of the anisotropic dynamic
susceptibility of CNTs as a function of momentum, length, and the tube
primitive indices. We intend to improve our perfect-cylindrical-conductor
simulation [1] by calculating the anisotropic dielectric tensor within a
tight-binding approach, providing
accurate binding energies and life
Collaborators:
Himadri S. Chakraborty (NW Missouri
State University, Maryville)
[1] M.
Zamkov, H.S. Chakraborty, A. Habib, N. Woody, U. Thumm, and P. Richard, Phys.
Rev. B 70, 115419 (2004).
[2] M.
Zamkov, N. Woody, B. Shan, H.S. Chakraborty, Z. Chang, U. Thumm, and P.
Richard, Phys. Rev. Lett. 93, 156803
(2004).
3. Laser-assisted collisions
Project scope: To investigate the effects a strong laser field has on
electron capture, emission, and level hybridization in ion-atom and
particle-surface interactions. These investigations are also intended to assist
in the planning of future experiments with crossed laser and pulsed particle
beams.
|
|
|
Fig. 1. Collision
scenario for a proton colliding at impact parameter b with an atomic hydrogen
target. For positive impact parameters, the projectile follows the rotating
circularly polarized laser field (“co-rotating” case). For negative impact
parameters, the projectile moves against the rotating electric field
(“counter-rotating” case). α denotes the angle between the collision
plane and the plane defined by the rotating laser electric field vector. |
|
Recent
progress: We
calculated cross sections for electron capture and emission in slow (keV)
prototype proton-hydrogen collisions in the presence of a strong 1064 nm
laser field. We first developed and applied a simplified 2D reduced
dimensionality model of the scattering system [1] in which the motion of the
active electron and the laser electric field vector are confined to the
scattering plane (Fig. 1). We then extended these 2D calculations by
propagating the full time-dependent 3D Schrödinger equation on a numerical
grid [2]. We examined the probabilities for electron capture and ionization
as a function of the laser intensity, the projectile impact parameter b, the
angle α between the collision plane, the plane in which the circularly
polarized laser electric field vector rotates, and the laser phase φ
that determines the orientation of the laser electric field with respect to
the internuclear axis at the time of closest approach between target and
projectile. Since the laser electric field breaks the cylindrical symmetry of
the collision system, our 3D calculations of laser-assisted capture and
ionization cross sections require the inclusion of a large number of
projectile trajectories. We found a relatively weak variation of the
laser-phase-resolved capture cross section on the angle α, such that our
2D and full 3D cross sections are qualitatively similar functions of φ. We
tested the accuracy of our 3D numerical wave function propagation calculation
by turning the laser field off and found agreement with known experimental
capture cross sections for p + H collisions. Both, laser-assisted ionization
and capture probabilities show a strong dependence on φ and on the
helicity of the circularly polarized laser light (Fig. 2). For intensities
above 2x1012 W/cm2, we predict a noticeable circular
dichroism in the capture probability for slow proton-hydrogen collisions that
persist after averaging over φ. Capture and electron emission
probabilities defer significantly from results for laser-unassisted
collisions. Ionization probabilities depend less sensitively on φ,
and their phase averages differ much less for co- and counter-rotating
collisions than the phase-averaged capture probabilities. For 1.21 keV
protons, the difference in the capture cross sections for co- and counter
rotating collisions at a laser intensity of 5x1013 W/cm2 amounts
to 40% in our 2D and to 15% in our 3D calculations. Our full 3D calculations
confirm evidence found in previous 2D calculations for a charge-resonance-enhanced-ionization
mechanism that may enable the measurement of φ. We predicted that laser
pulses with lengths of a few nanoseconds and intensities of about 1012
W/cm2 and higher would allow for the experimental verification of
the calculated dichroism in the capture probability. |
|
|
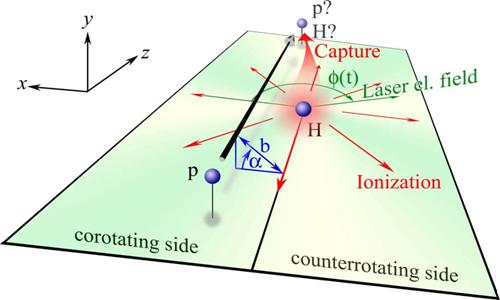
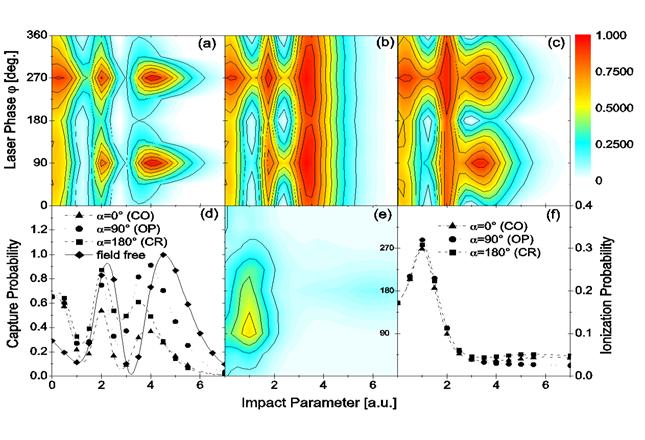
Fig. 2. (a) - (c) Contour plots of the electron capture probability and (e) the ionization probability as a function of impact parameter and laser
phase φ for (a, e) co-rotating (CO), (b) off-plane with α = ± 90 degrees (OP), and (c) counter-rotating (CR) collisions. Also shown are the phase-averaged
results for (d) capture and (f) ionization, together with the field-free probabilities.
|
Future plans: …are largely dependent on the experimental progress
in synchronizing particle beams with intense laser pulses. If encouraged by
future experiments, we will attempt to refine our studies, e.g., by including
electronic correlation in two-electron projectile ions, by providing
sublevel-specific excitation and angle-resolved electron-emission cross
sections, and by adjusting collision and laser parameters to the experiment.
Collaborator: Thomas Niederhausen (KSU, now at University of Madrid, Spain)
[1] T. Niederhausen, B.
Feuerstein, and U. Thumm, Phys. Rev. A 70, 023408 (2004).
[2] T. Niederhausen and U.
Thumm, Phys. Rev. A 73, 041404(R) (2006).
4. Highly correlated negative-ion resonances in
photodetachment and electron scattering processes
Project scope: (i) To explore the effect of electronic
correlation in negative ions on photo-detachments and electron-scattering
processes and (ii) to provide basic data (e.g., scattering lengths) for the
modeling of ultracold collisions between alkali atoms and the formation of
alkali dimers in atomic traps.
Recent progress: In the field of electron‑atom interactions,
alkali‑metal atoms and noble gases are frequently chosen targets for
detailed experimental and theoretical studies, owing both to their relative
theoretical simplicity and to the relative ease with which they can be handled
experimentally. The heavier targets,
like rubidium, cesium, or francium allow for the exploration of relativistic
effects, which are too small to be easily observed in lighter atoms.
For the electron‑cesium
system, two multiplets of narrow shape resonances (with widths of a few meV)
are of particular interest. We have shown [1] that these resonances are influenced
by both (two‑electron) core-polarization and relativistic effects. The former convert the 3PoJ
negative-ion states from bound states to resonances. The latter add fine‑structure splitting
and finite auto-ionization widths to 3PeJ
states that in LS coupling are strictly uncoupled to the adjacent continuum
(Fig. 1). Our calculations predict the same resonances to occur in Rb‑ and Fr‑ [2].
We have identified and
characterized a large number of scattering resonances in elastic, inelastic,
angle-differential, and total electron-scattering cross sections for Rb, Cs,
and Fr (atomic) targets. Our results for 3Po and 3Fo
shape resonances and 3Pe, 1Po, and 1D0
Feshbach resonances of Rb-, Cs-, and Fr-
negative ions are in agreement with available experimental data [2]. We have
calculated the 3Se and 1Se
scattering lengths in ultraslow collisions of electrons with ground state Rb,
Cs, and Fr atoms. These calculations are based on a new relativistic effective-range
theory that allows us to extrapolate eigenphases that are provided by relativistic
Dirac R-matrix calculations to zero energy [6]. Recently, our scattering
lengths have contributed to the prediction of a new class of highly excited,
trilobite-shaped states of Rb2 dimers [7].
|
Fig. 1. Total elastic (thick solid line) and inelastic cross sections: ns1/2 → np1/2 (dotted line), ns1/2 → np3/2 (short dashed line), ns1/2 → (n-1)d3/2
(long dashed line) and ns1/2
→ (n-1)d5/2 (thin
solid line) for e- + Rb, Cs, and Fr scattering (n = 5 for Rb, 6 for Cs, and 7 for
Fr). The stars show the two-state close-coupling results of Fabrikant [3],
the circles and squares represent the measured total scattering cross
sections of Visconti et al. [4] and
of Jaduszliwer and Chan [5]. For both experiments, the size of the symbols
indicates the total error in the cross sections. The arrows point to the
position of a 3Fo resonance. The insets give the
elastic cross section for energies below 0.5 eV and show the profile of the 3Po
shape resonances. |
|
Fig.
2. Angle-integrated
photodetachment cross sections of Cs- near the detachment
threshold. Dotted curve: S=0 contribution only; dashed curve: total cross section for a cutoff parameter
in the induced dipole potential rc = 2.1271; solid curve: same
result for rc=2.1294. Circles show the experimental data of [8]. The inset shows
our calculated S = 1 contribution to the detachment cross section (thick
solid curve) compared to the background- subtracted measurement (dots) [8]. The sizes of the
circles and dots indicate the experimental error. |
Future plans: The dipole allowed J=1 term of the aforementioned 3PoJ
resonances was confirmed in measured photoelectron spectra [8] (Fig.
2). We plan to extend our relativistic calculations towards the
consistent description of photo-processes within the Dirac R-Matrix approach [9].
These calculations will provide reliable absolute cross sections for the photodetachment
of atoms and ions, as well as atomic alignment and coherence parameters.
We intend to study the decay and photodetachment of
alkali-metal negative ions in the presence of strong external fields. Placing atoms or ions in a combination of
laser and static fields allows the manipulation of the detachment process and
to observe basic quantum mechanical effects such as multi-path interference. Reflection
of the detached electron wave packet from the potential barrier created by a
static external field leads to interference with the "direct"
(downfield) emission path. Observation of these interferences provides detailed
information about the electron-atom scattering amplitude at laser
resolution. This project will build on
our experience with Dirac R-matrix applications and will lead to a new set of
computer programs to investigate photo-processes of stable or resonant, non-Coulombic
systems with two active electrons in an external field. The provided basic
atomic data are relevant for applications in chemistry, physics, and
engineering.
Collaborators: Cristian Bahrim (Lamar University, Texas) and Ilya
Fabrikant (University of Nebraska, Lincoln)
[1] U. Thumm and
D.W. Norcross, Phys. Rev. Lett. 67,
3495 (1991); C. Bahrim and U. Thumm, Phys. Rev. A 61, 022722 (2000).
[2] C. Bahrim, U. Thumm, and I.I. Fabrikant, Phys. Rev. A 63, 042710 (2001); C. Bahrim and U.
Thumm, Phys. Rev. A 64, 022716 (2001).
[3] I. Fabrikant, Phys. Lett. A 58, 21 (1976).
[4] P.J. Visconti, J.A. Slevin, and
K. Rubin, Phys. Rev. A 3, 1310
(1971).
[5] B. Jaduszliwer and Y.C. Chan,
Phys. Rev. A 45, 197 (1992).
[6] C. Bahrim, U. Thumm, and I.I. Fabrikant, J. Phys.
B 34, L195 (2001).
[7] C. H. Greene, A. S. Dickenson, and H. R.
Sadeghpour, Phys. Rev. Lett. 85,
2458 (2000).
[8] M. Scheer et al., Phys. Rev. Lett. 80, 684 (1998).
[9] C. Bahrim,
Updated
9/28/11