Towards the Simulation of Bulk Properties via Molecular Dynamics
Mairead Heiger
Kansas State University
Physics Major, minors in economics and mathematics
Mentored by Dr. Jeremy Schmit
The focus of this project is on accurate modelling of salt conditions near biological macromolecules, such as proteins or DNA. The function and structure of these macromolecules are very sensitive to salt content, and the local environment in which they are found, such as the cytoplasm, is often an ionic solution. The mobile ions in the ionic solution will cluster around a charged macromolecule in a process known as electrostatic screening, resulting in a dynamic local salt concentration that can differ greatly from the bulk concentration. Many biomolecular simulations, however, fix their salt content, and so may not accurately model the actual salt content and the fluctuations in salt content in the environment near the protein. Accurate modelling of salt effects is important to understanding molecular dynamics, as well as to pharmacological design: Many new drugs are ligands, small molecules that bind to protein and are known to be modulated by salt effects (Ross 2017).
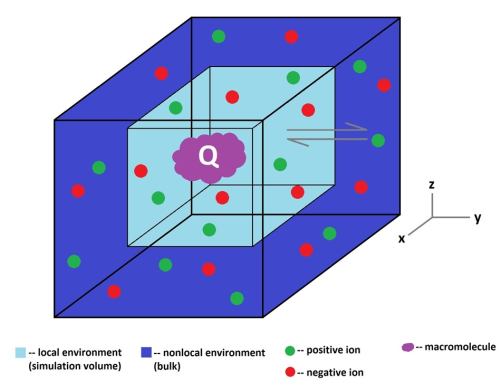
Figure 1: Ions (green and red) move in and out of a simulation volume (light blue) containing a protein (purple). The effects on the free energy of the system of these fluctuations in ion content within the simulation volume are the subject of this project.
This project used analytic theory to develop a model of independent fluctuations in salt content near a charged macromolecule. The grand canonical partition function, which relates the microscopic quantities of the system, like individual energies, to the macroscopic quantities, like pressure or free energy, was used to describe a system in which cations and anions fluctuated independently of one another.
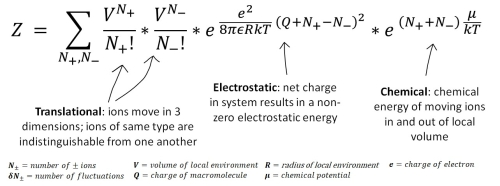
Figure 2: The partition function for the grand canonical ensemble of the model of independent fluctuations in ion content.
We evaluated this expression and, since the free energy is proportional to the natural log of the partition function, we used this to find an expression for the free energy, F, as a function of N± (the number of ions), 𝛿N± (the number of fluctuations), Q (the charge of the macromolecule), V (the volume of the local environment considered), and c0, the bulk concentration. Then, using SLTCAP, a method for calculating the number of ions in a volume perturbed by a charge that Dr. Schmit had developed before the REU, we substituted for N±. The resulting expression depended only on 𝛿N±, Q, V, and c0. We then evaluated this expression numerically for different combinations of fluctuations.
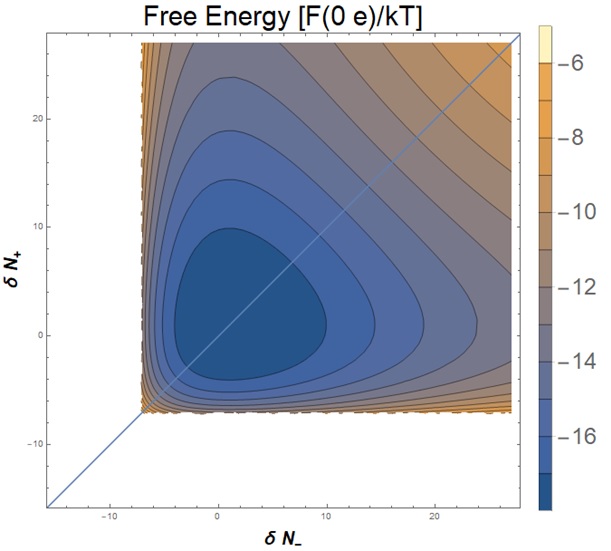
Figure 3: The free energy of a system with no background charge as a function of 𝛿N+ and 𝛿N-.
Using this expression for the free energy of the system, we could model simple processes of macromolecules. Here, we represent the folding of a protein with two charged parts. Two separate boxes (volumes) of charge Q (the charged parts are neutral to each other due to screening layer effects) become one box of charge 2Q (when the protein folds, the charged parts are close enough to interact) and one with zero charge.
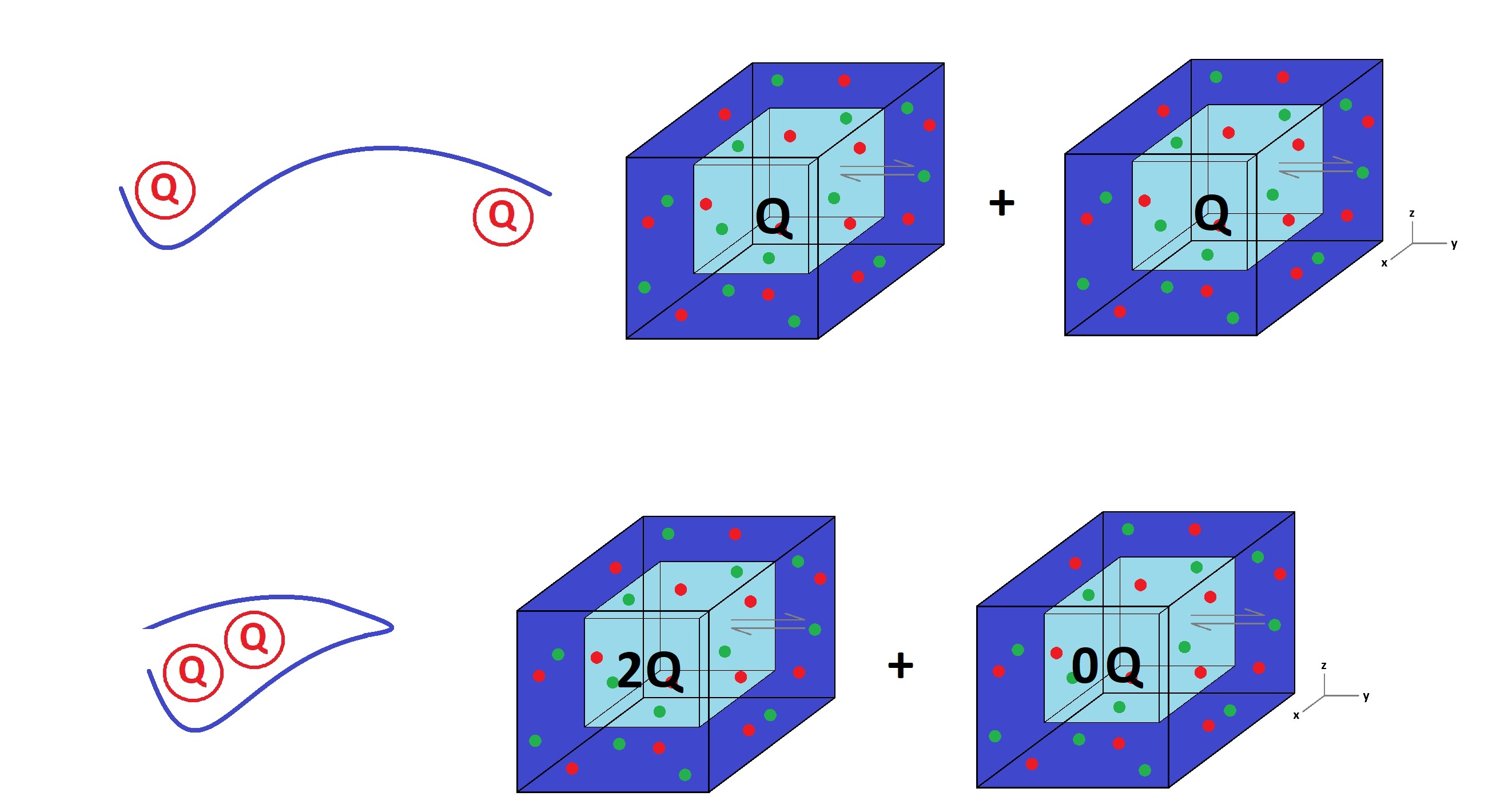
Figure 4: Modelling a protein folding with the model of independent salt fluctuations.
We then compared the change in free energy, ΔF, from [2F(Q)] (unfolded) - [F(2Q)+F(0)] (folded) with fluctuations to ΔF without fluctuations. We are still analyzing these results, and hope to implement this model in a simulation.
References
[1] Ross, Gregory A., et al. ''Biomolecular Simulations under Realistic Macroscopic Salt Conditions.'' Preprint Ahead of Submission, 2017, doi:10.1101/226001.; OpenStax, Biology. OpenStax CNX. May 13, 2015 http://cnx.org/contents/185cbf87-c72e-48f5-b51e-f14f21b5eabd@9.85
[2] Schmit, Jeremy D., et al. ''SLTCAP: A Simple Method for Calculating the Number of Ions Needed for MD Simulation.'' Journal of Chemical Theory and Computation, vol. 14, no. 4, 2018, pp. 1823–1827., doi:10.1021/acs.jctc.7b01254.
Acknowledgments
Thanks to Dr. Schmit, Nelson Ramallo, and other group members for their support and insight throughout this project, as well as to Kansas State, the NSF, and the REU program.