Surface Anharmonicity -- In this effort we
use two techniques to calculate the line width and frequency shift of phonons
at metallic surfaces. First, we use perturbation theory, determining the
perturbation from the Taylor-series expansion of an analytic representation
of a reliable semi-empirical embedded-atom potential. Second, we use molecular
dynamics simulations based on embedded-atom interaction potentials. Here
line width and frequency are determined from a phonon spectral density
calculated via a velocity-velocity correlation function.
The anharmonicity
of vibrations in solids is well recognized and is required to account for
many macroscopic properties. Using molecular dynamics simulations,
we have calculated some of these properties as functions of temperature:
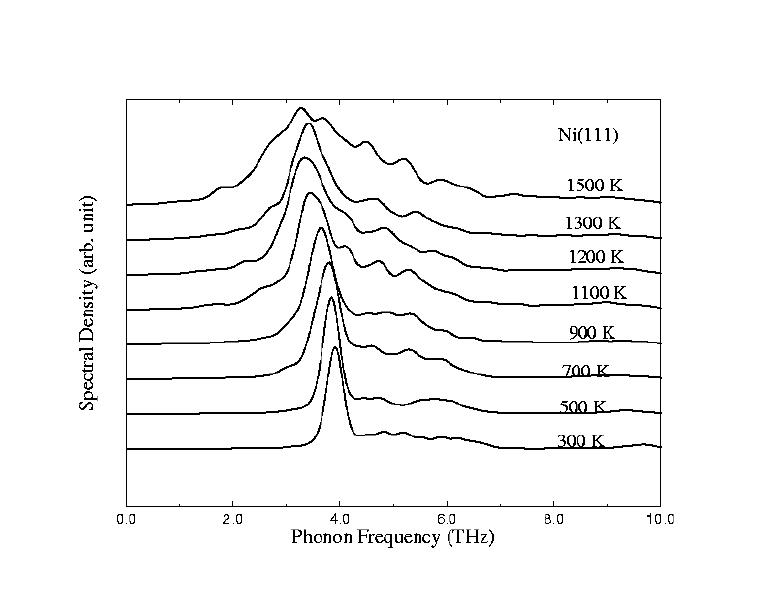
| Phonon spectroscopies and lifetimes, as shown in figure (1) for Ni(111) from room temperature up to 0.9 Tm (where Tm is the melting temperature) | |
|
|
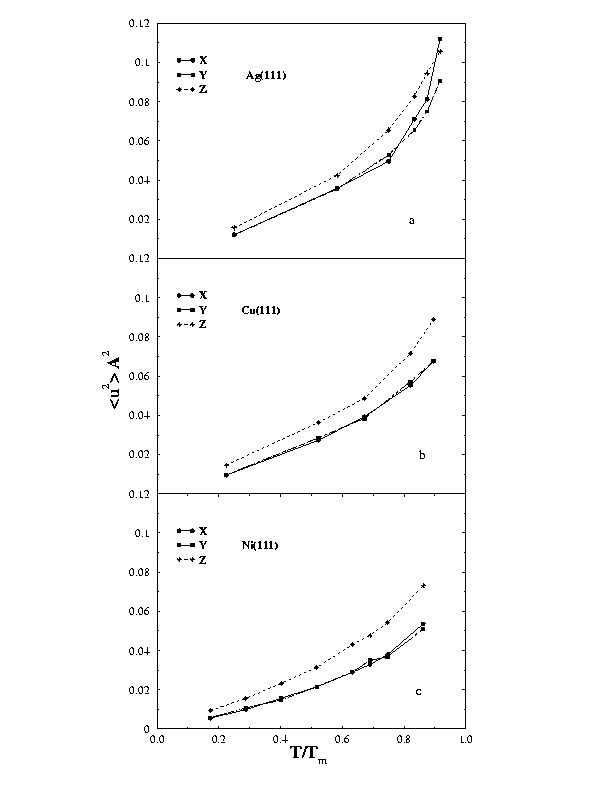
In figure (2), we calculated the mean squared vibrational amplitudes for Ag(111), Cu(111) & Ni(111) up to temperatures near the melting point. |
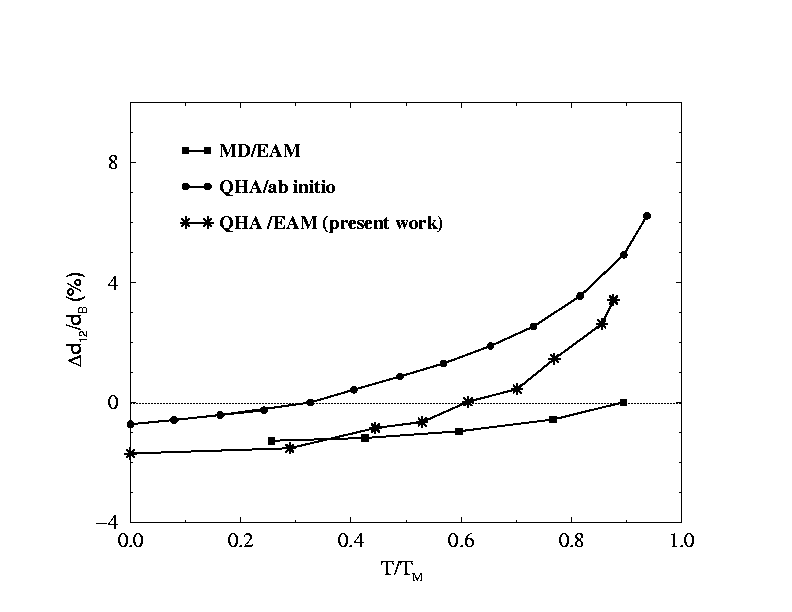
| Finally, we calculated the thermal expansion of the metal surfaces Ag(111), Cu(111) and Ni(111). In figure (3) we show the results of our calculations using molecular dynamics with a potential from the embedded atom method (EAM). With two calculations using the quasi-harmonic approximation one of whose potentials came from ab initio calculations, the other from EAM. |
|