-
Computational studies of adsorption, diffusion and reactions of atoms and molecules on metal oxide surfaces.

Focus of this project is on microscopic mechanisms of oxidation of carbon monoxide on Cu2O surfaces and nanoclusters and oxidation of ammonia (NH3) on RuO2(110) surface. Our calculations result in adsorption energies, as well as, diffusion and reaction activation barriers. These quantities provide important information about reaction energetics and pathways and determine the rate limiting reaction steps. The valence charge density and densities of electronic states obtained from such calculations tells us about character of chemical bonding between reactants and surface at various stages of the reaction and help to rationalize the revealed catalytic effects. By introducing the chemical potentials into the free energy of the system we can consider the oxide surface in equilibrium with gaseous reactants and calculate from first principles the surface structure (composition and geometry) as a function of temperature and pressure of oxygen. Finally, a set of activation barriers for elementary catalytic processes obtained from first principles calculations will be used in kinetic Monte Carlo simulations which result in a consistent description of surface reaction including the temperature and pressure dependence of reaction rate.
-
Rationalization of the promotion and poison effects of co-adsorbed atoms on reactivity of metal catalyst surfaces.
One part of the project is the first principles study of the effect of adsorption of carbon and sulfur (poisons) or alkali metals (promoters) on the electronic structure of flat and stepped metal catalyst (Pd, Cu, Ni) surfaces. Another part is a computational study of the energetics of the adsorption, diffusion and reactions of CO and NH3 on metal catalyst surfaces with and without co-adsorbed promoters or poisons. Analysis of the electronic structure of the systems in initial, transition, final and some intermediate states will help us to understand the mechanisms of influence of co-adsorbed poisoning or promoting atoms on reaction activation barriers.
-
Adsorption and reactivity of NO on RuO2(110) surface.
 We
report here results of density functional theory (DFT) calculations in the
generalized gradient approximation (GGA) using the Perdew-Wang
functional for exchange-correlation energies on the energetics of
adsorption, oxidation and dissociation of NO on
We
report here results of density functional theory (DFT) calculations in the
generalized gradient approximation (GGA) using the Perdew-Wang
functional for exchange-correlation energies on the energetics of
adsorption, oxidation and dissociation of NO on stoichiometric RuO2(110) surface and compare them to experimental
data. We find that NO adsorbs on top of the undercoordinated Ru with a
tilted axis and the adsorption energy changes from 1.65 eV for half
monolayer coverage to 1.43 eV for a full monolayer coverage. Unlike CO, NO
oxidation was not observed in the surface and we find that it can be
explained by the
difference in their electronic structure, i.e, different locations of
antinbonding states. For NO dissociation, the relevant calculated energy
barrier is 2.3 eV and a stable final state is not found suggesting no
dissociation on the surface.
stoichiometric RuO2(110) surface and compare them to experimental
data. We find that NO adsorbs on top of the undercoordinated Ru with a
tilted axis and the adsorption energy changes from 1.65 eV for half
monolayer coverage to 1.43 eV for a full monolayer coverage. Unlike CO, NO
oxidation was not observed in the surface and we find that it can be
explained by the
difference in their electronic structure, i.e, different locations of
antinbonding states. For NO dissociation, the relevant calculated energy
barrier is 2.3 eV and a stable final state is not found suggesting no
dissociation on the surface.
Some recent results:
-
Based on first principles calculations, we find alkali adsorbates to induce a dramatic enhancement of
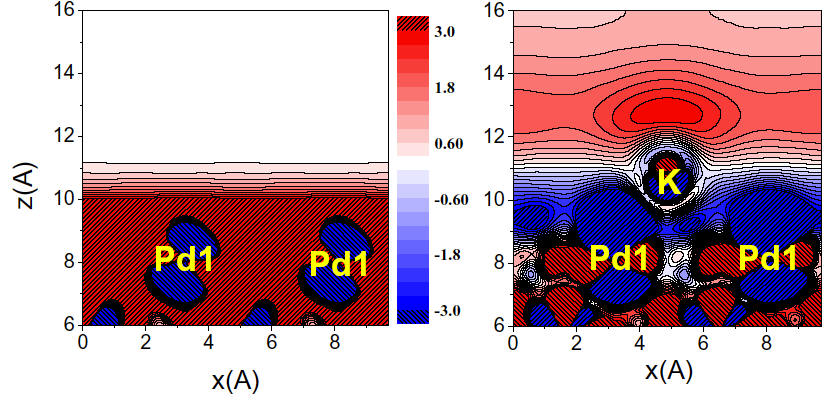 the electronic polarizability of metal surfaces extending it
several angstrom into the vacuum {Phys.
Rev. Lett. Accepted for publication}.
We show that this effect is responsible for the experimentally observed
softening of vibrational modes of molecules co-adsorbed with alkalis on
metal surfaces. Our results also suggest that the enhanced polarizability
can be a driving force for well-known promotion effect in heterogeneous
catalysis (increase of surface reactivity upon alkali adsorption).
the electronic polarizability of metal surfaces extending it
several angstrom into the vacuum {Phys.
Rev. Lett. Accepted for publication}.
We show that this effect is responsible for the experimentally observed
softening of vibrational modes of molecules co-adsorbed with alkalis on
metal surfaces. Our results also suggest that the enhanced polarizability
can be a driving force for well-known promotion effect in heterogeneous
catalysis (increase of surface reactivity upon alkali adsorption).
-
Unidirectional adsorbate motion on a high-symmetry surface: ``Walking'' molecules can stay the
 course
{Phys.
Rev. Lett. 95, 166101 (2005)
see also Physics Today
58, No.12, p. 9 (2005),
Physics News Update (AIP) No751 #2 (2005). This is also in the list of
the Top Physics Stories for 2005,
Physics News Update (AIP) No757 (2005)}.
STM experiment performed in University of California, Riverside reveals
unusual diffusive motion of an organic molecule along a straight line on
the Cu(111) surface, and first principles calculations of potential energy
surface for diffusion of the molecule explain the mechanism of this
phenomenon.
course
{Phys.
Rev. Lett. 95, 166101 (2005)
see also Physics Today
58, No.12, p. 9 (2005),
Physics News Update (AIP) No751 #2 (2005). This is also in the list of
the Top Physics Stories for 2005,
Physics News Update (AIP) No757 (2005)}.
STM experiment performed in University of California, Riverside reveals
unusual diffusive motion of an organic molecule along a straight line on
the Cu(111) surface, and first principles calculations of potential energy
surface for diffusion of the molecule explain the mechanism of this
phenomenon.
-
Adsorption, diffusion and dissociation of NH3 on steps and terraces of Ni and Pd surfaces J.
 Chem. Phys. 123, 204716 (2005). Based on the first principles
calculations we show that the activation barrier for NH3
dissociation on metal surfaces is much lower for the molecule adsorbed on
monoatomic steps than on terrace. In contrast to the Ni surface case, for
Pd surface the activation barrier is found to be higher than disorption
energy even if the molecule is adsorbed at the step. This explains the
experimental finding, which shows that NH3 decomposition rate
is much higher on Ni than on Pd.
Chem. Phys. 123, 204716 (2005). Based on the first principles
calculations we show that the activation barrier for NH3
dissociation on metal surfaces is much lower for the molecule adsorbed on
monoatomic steps than on terrace. In contrast to the Ni surface case, for
Pd surface the activation barrier is found to be higher than disorption
energy even if the molecule is adsorbed at the step. This explains the
experimental finding, which shows that NH3 decomposition rate
is much higher on Ni than on Pd.
-
As coverage of carbon atoms adsorbed on the Ni(001) surface exceeds 1/3 of monolayer (ML), so
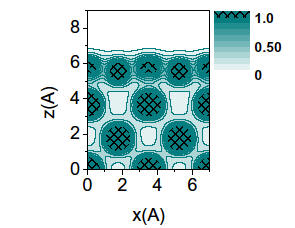 -called
“clock reconstruction” of the surface
is observed in experiments. The results of our calculations of the
geometric structures of the system with the coverage of 1/4 and 1/2 ML are
in a very good quantitative agreement with the experimental findings.
Furthermore, we obtain the reconstruction pathway and energetics. Analysis
of the calculated valence charge density distribution in the system allows
us to explain the mechanism of the reconstruction. {Phys.
Rev. 72,155423
(2005)}); also
selected for the October 31, 2005 issue of Virtual Journal of Nanoscale
Science & Technology (AIP, APS)
http://www.vjnano.org/.
-called
“clock reconstruction” of the surface
is observed in experiments. The results of our calculations of the
geometric structures of the system with the coverage of 1/4 and 1/2 ML are
in a very good quantitative agreement with the experimental findings.
Furthermore, we obtain the reconstruction pathway and energetics. Analysis
of the calculated valence charge density distribution in the system allows
us to explain the mechanism of the reconstruction. {Phys.
Rev. 72,155423
(2005)}); also
selected for the October 31, 2005 issue of Virtual Journal of Nanoscale
Science & Technology (AIP, APS)
http://www.vjnano.org/.
-
Effect of adsorption of C and S on geometric and electronic structure of stepped Pd surfaces –
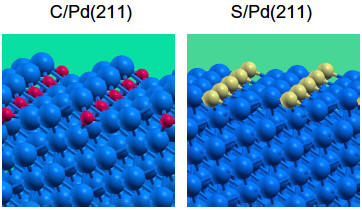 poison reactivity {Phys.
Rev. B 70, 155410 (2004)}; We have performed first principles
calculation of C and S adsorption energies for various possible adsorption
sites on Pd(211) surface to reveal the preferred adsorption geometries.
We find that hybridization between C p-states and neighboring Pd
d-states leads to the formation of strong covalent C-Pd bonds, while
S-Pd bonding has predominantly ionic character. The depletion of the
electronic density of states of the Pd surface atoms at the Fermi level
suggests a poisoning effect of C on catalytic activity of the surface.
poison reactivity {Phys.
Rev. B 70, 155410 (2004)}; We have performed first principles
calculation of C and S adsorption energies for various possible adsorption
sites on Pd(211) surface to reveal the preferred adsorption geometries.
We find that hybridization between C p-states and neighboring Pd
d-states leads to the formation of strong covalent C-Pd bonds, while
S-Pd bonding has predominantly ionic character. The depletion of the
electronic density of states of the Pd surface atoms at the Fermi level
suggests a poisoning effect of C on catalytic activity of the surface.
-
Our calculations help to understand unusual reflection anisotropy spectra obtained for nanostructured copper surfaces by our colleagues experimentalists from Switzerland {Phys. Rev. Lett. 90, 177402 (2003)}.
-
Our collaborators, experimentalists from Germany, show that oxygen works as surfactant promoting 2D Ni film growth on the Cu(001) surface. During the growth O atoms are found to float up and stay on the film surface that is critical for a surfactant. Our calculations reproduce this effect and suggest a mechanism for that floating {Surf. Sci. 531, 53 (2003)}.
-
Studying so-called “missing row reconstruction” of the Cu(100) surface upon oxygen adsorption we have found that the long-range electrostatic interaction between the surface and adsorbates is the main driving force for this phenomenon. {Phys. Rev. Lett. 89, 116101 (2002)} .