GREETINGS!!!!!!!
My
name is Alex Olinger, and I had the pleasure of working with Dr. Amit
Chakrabarti's group during this summer at the K-State physics REU. My
project was to simulate and study the aggregation of globular protein monomers
in solution. The codes I used were
written in Fortran90. The particles in my simulation are on the nanoscale,
having a diameter of 3-4 nm, and were under a random force which is described
through Brownian Dynamics. In my simulation, the proteins are represented
by spheres with a uniform surface charge distribution. Since there is a
uniform charge distribution each monomer has a fluctuating dipole moment which
causes an attractive Van der Waals force between the monomers. This attractive
potential is represented by a modified Lennard-Jones potential for hard
spheres. The charge of the monomers also
creates a repulsive Coulombic force. However, by effectively representing
salt in the solution containing the monomers, I can screen the Coulombic
potential between the monomers by increasing the concentration of salt in
solution which affects the number of negatively charged ions in solution. The
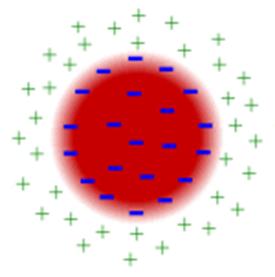
negatively charged ions are attracted to the proteins, resulting in the
creation of a neutral barrier around the protein. See figure (1). I
used the Yukawa potential for hard spheres to model this potential. I can
also effect the aggregation of the proteins by changing the volume fraction
(comparison of the number of particles and the size of my system) of my system
and the temperature of my system. In my simulations, the number of
particles and the temperature of the system remain constant. The
condition of constant temperature was used due to the relatively constant
temperature of the human body.
Figure
1